

Muscarinic receptor regulates extracellular signal regulated kinase by two modes of arrestin binding
- PMID: 28652372
- PMCID: PMC5514713
- DOI: 10.1073/pnas.1700331114
Muscarinic receptor regulates extracellular signal regulated kinase by two modes of arrestin binding
Abstract
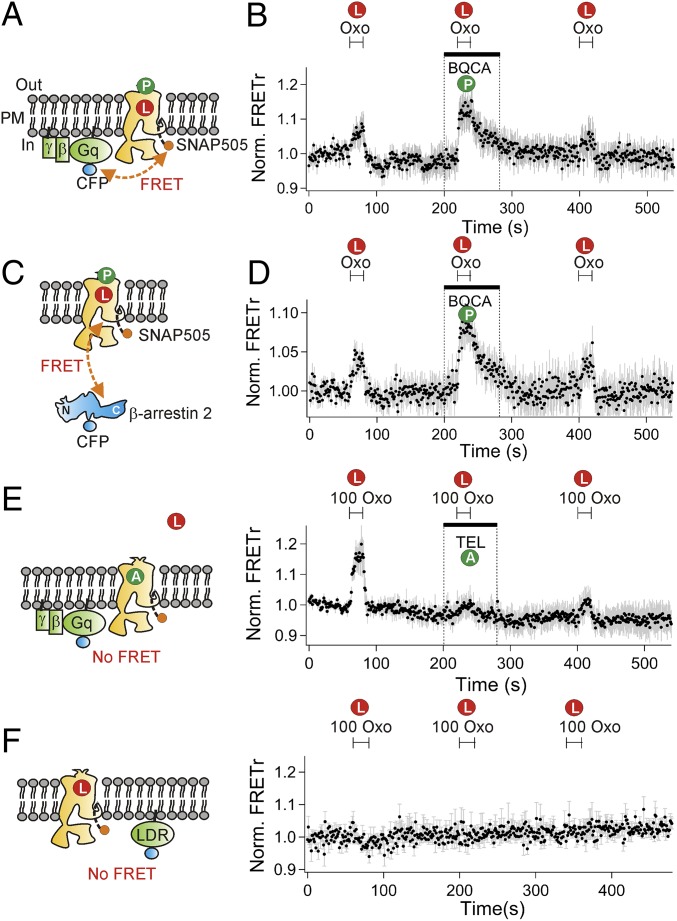
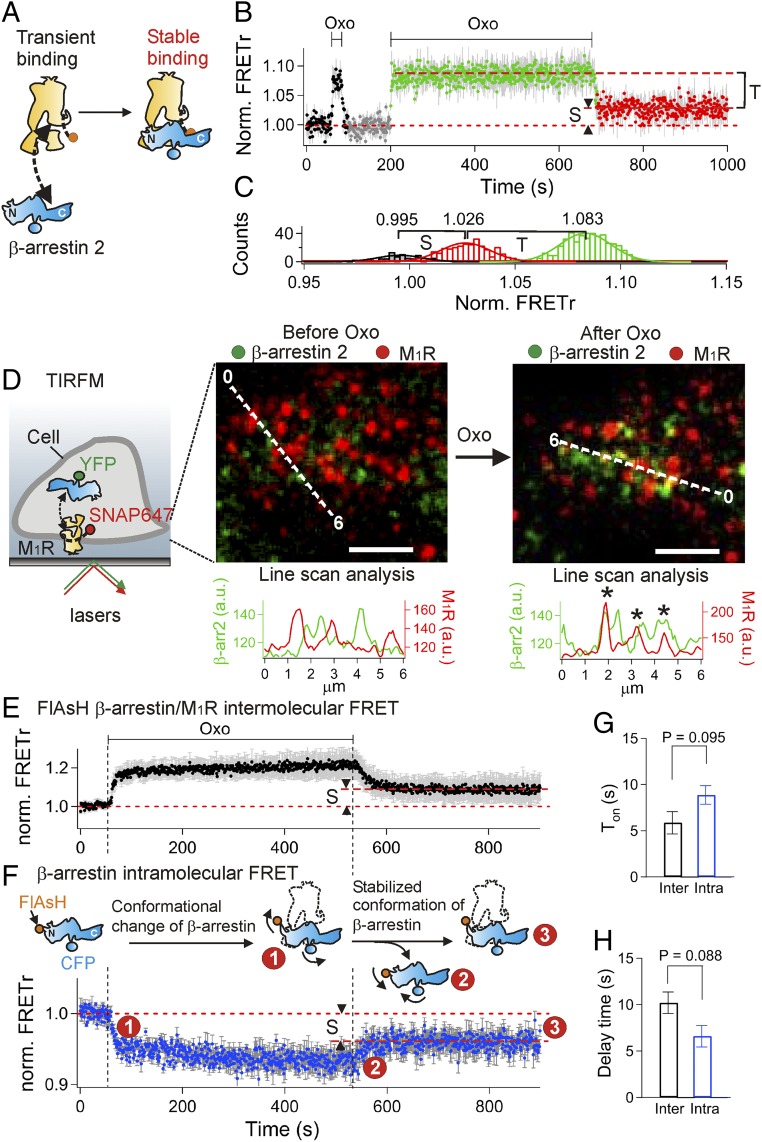
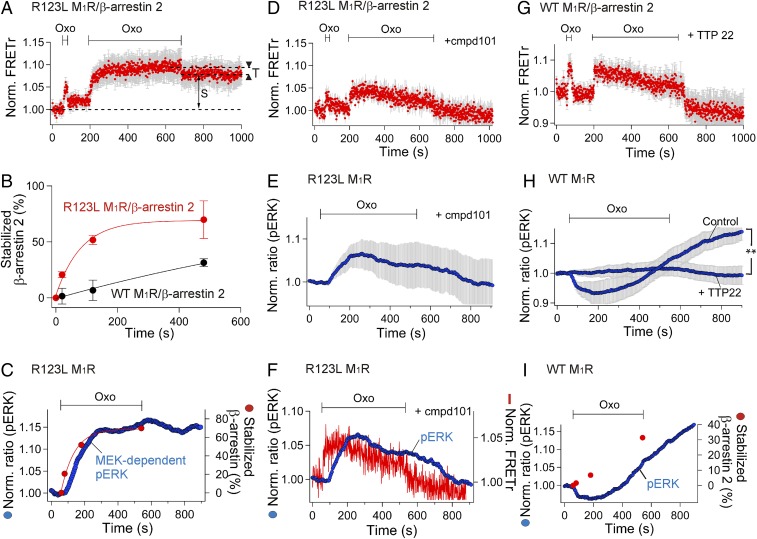
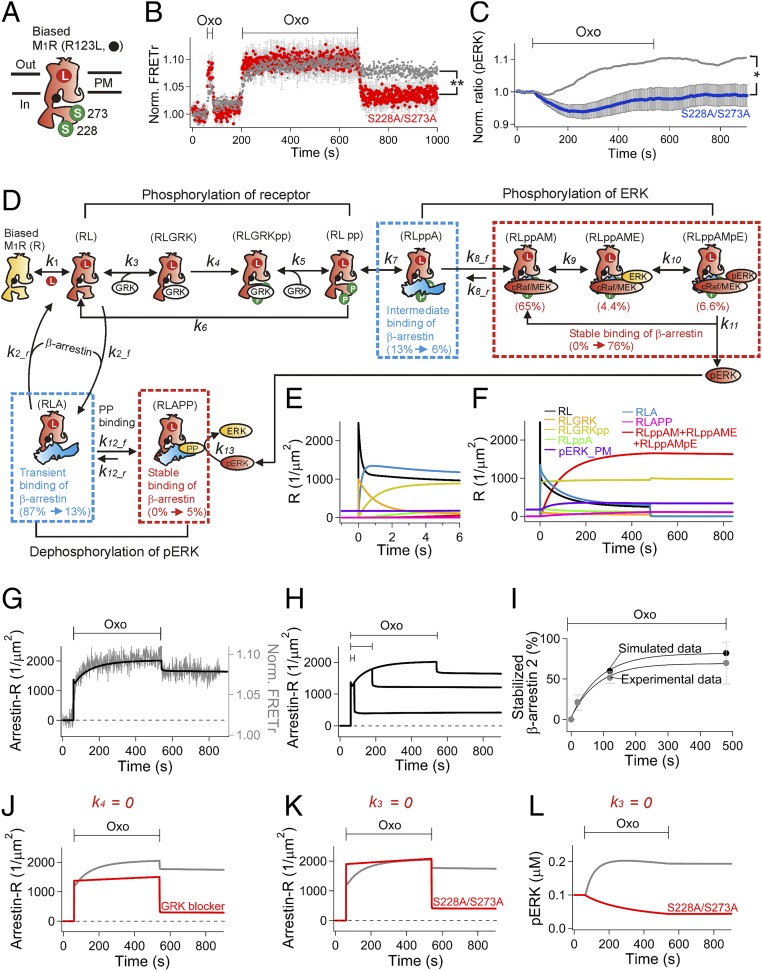
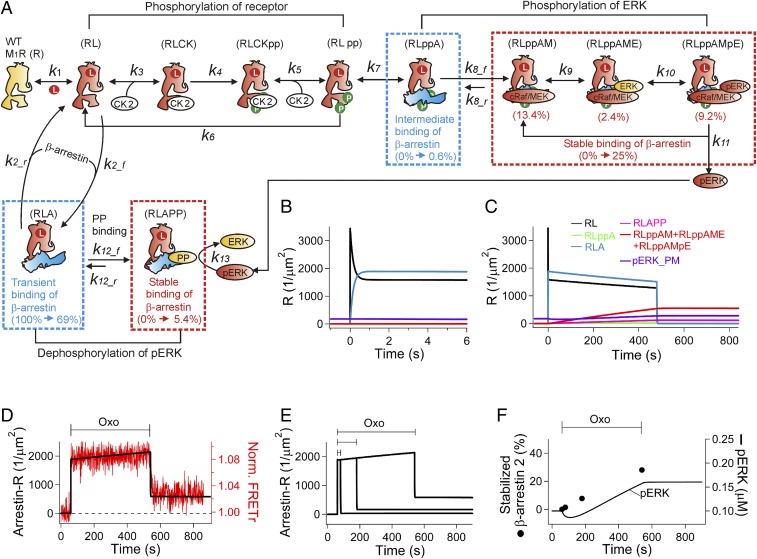
Binding of agonists to G-protein-coupled receptors (GPCRs) activates heterotrimeric G proteins and downstream signaling. Agonist-bound GPCRs are then phosphorylated by protein kinases and bound by arrestin to trigger desensitization and endocytosis. Arrestin plays another important signaling function. It recruits and regulates activity of an extracellular signal-regulated kinase (ERK) cascade. However, molecular details and timing of ERK activation remain fundamental unanswered questions that limit understanding of how arrestin-dependent GPCR signaling controls cell functions. Here we validate and model a system that tracks the dynamics of interactions of arrestin with receptors and of ERK activation using optical reporters. Our intermolecular FRET measurements in living cells are consistent with β-arrestin binding to M1 muscarinic acetylcholine receptors (M1Rs) in two different binding modes, transient and stable. The stable mode persists for minutes after agonist removal. The choice of mode is governed by phosphorylation on key residues in the third intracellular loop of the receptor. We detect a similar intramolecular conformational change in arrestin in either binding mode. It develops within seconds of arrestin binding to the M1 receptor, and it reverses within seconds of arrestin unbinding from the transient binding mode. Furthermore, we observed that, when stably bound to phosphorylated M1R, β-arrestin scaffolds and activates MEK-dependent ERK. In contrast, when transiently bound, β-arrestin reduces ERK activity via recruitment of a protein phosphatase. All this ERK signaling develops at the plasma membrane. In this scaffolding hypothesis, a shifting balance between the two arrestin binding modes determines the degree of ERK activation at the membrane.
Keywords: ERK; GPCR; arrestin; muscarinic receptor; receptor kinase.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Doan T, Mendez A, Detwiler PB, Chen J, Rieke F. Multiple phosphorylation sites confer reproducibility of the rod’s single-photon responses. Science. 2006;313:530–533. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
