

Decoding the Mechanism of Intramolecular Cu-Directed Hydroxylation of sp3 C-H Bonds
- PMID: 28654755
- PMCID: PMC5792191
- DOI: 10.1021/acs.joc.7b01069
Decoding the Mechanism of Intramolecular Cu-Directed Hydroxylation of sp3 C-H Bonds
Abstract
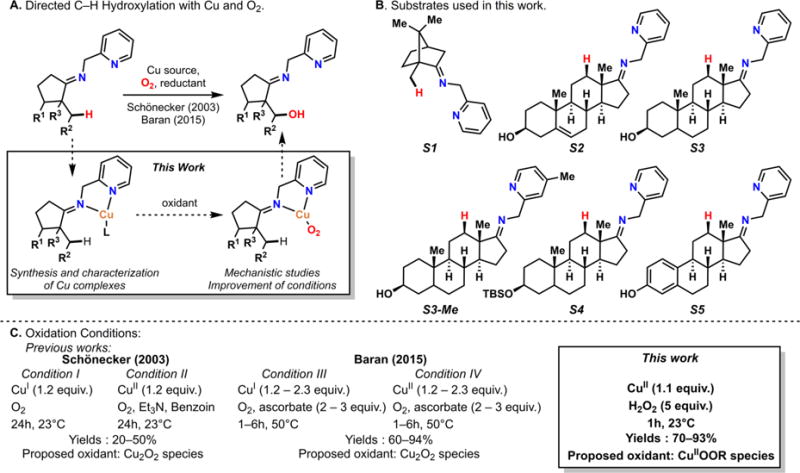
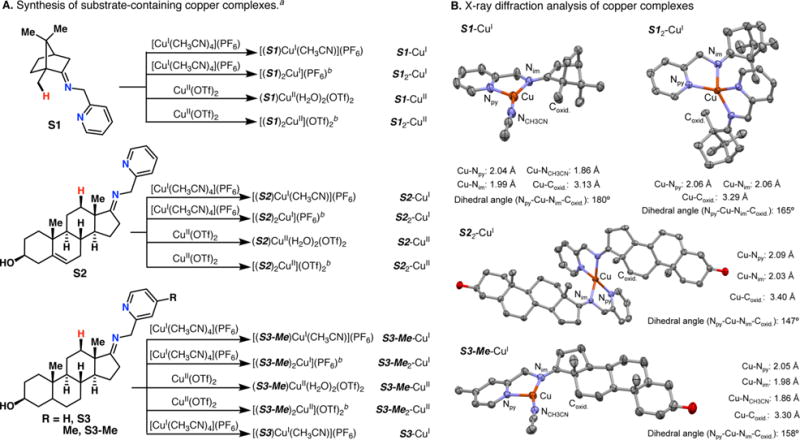
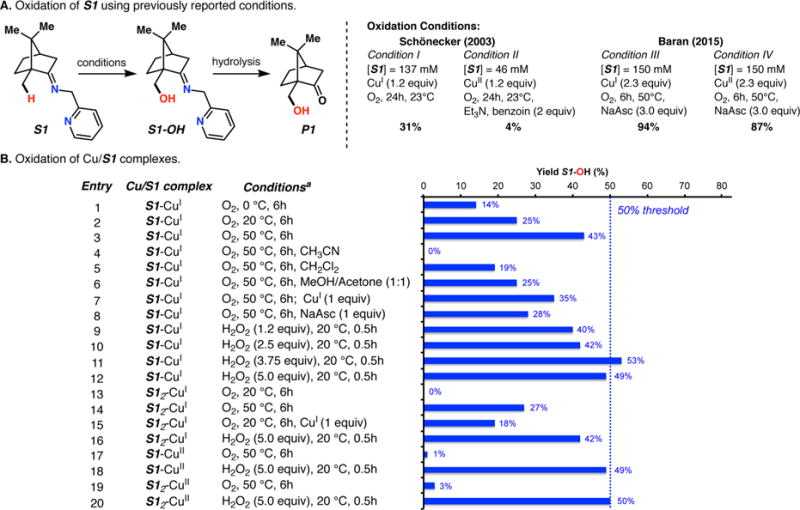
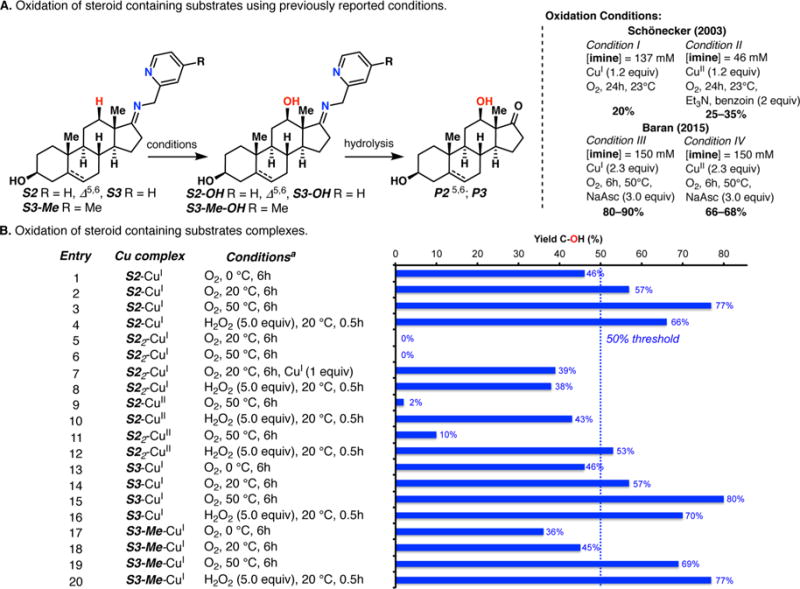
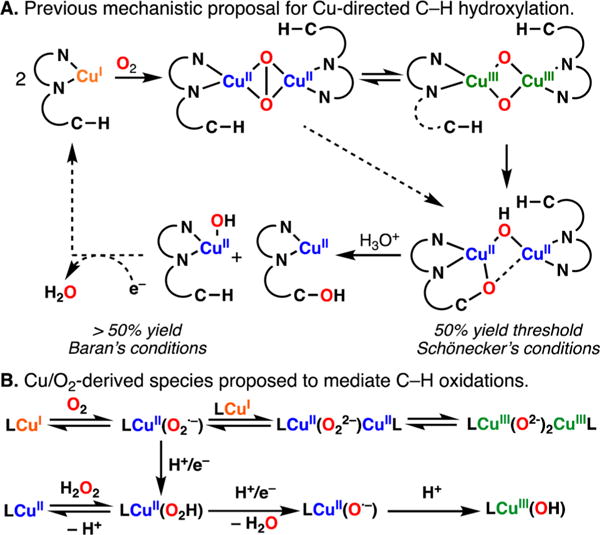
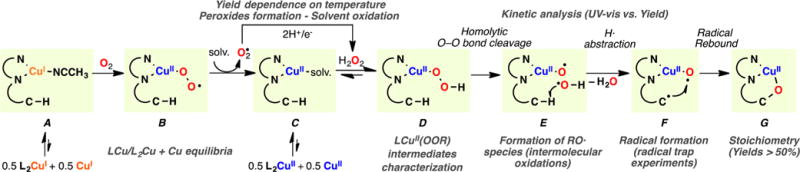
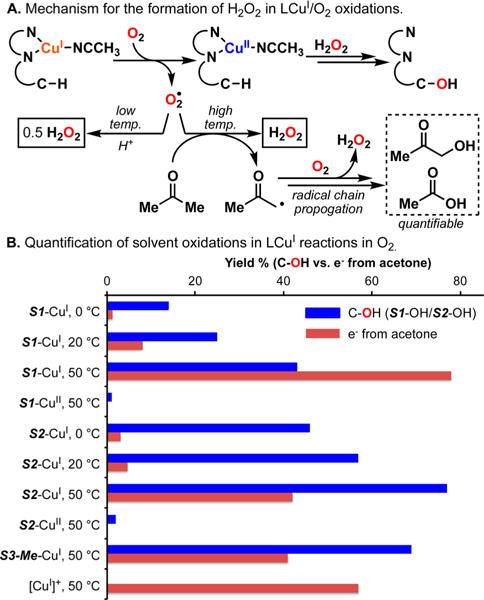
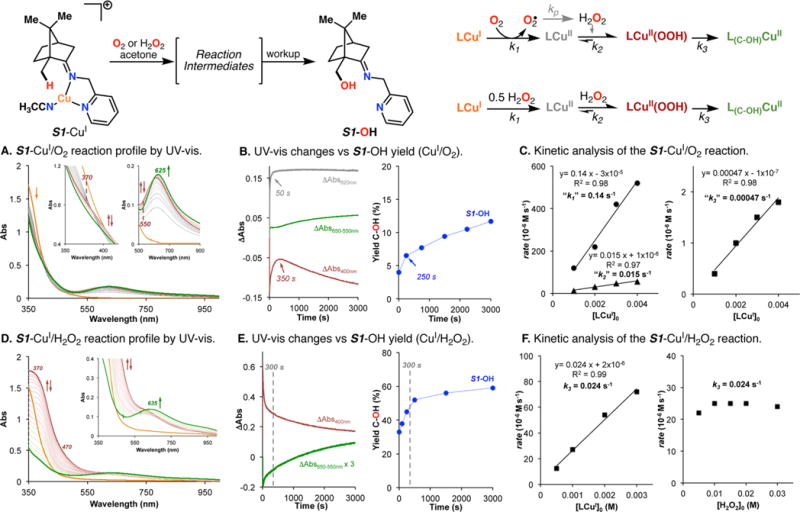
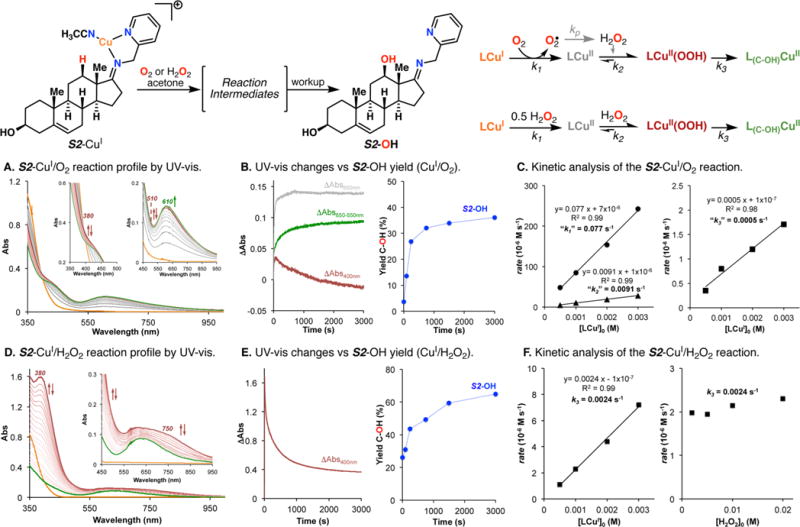
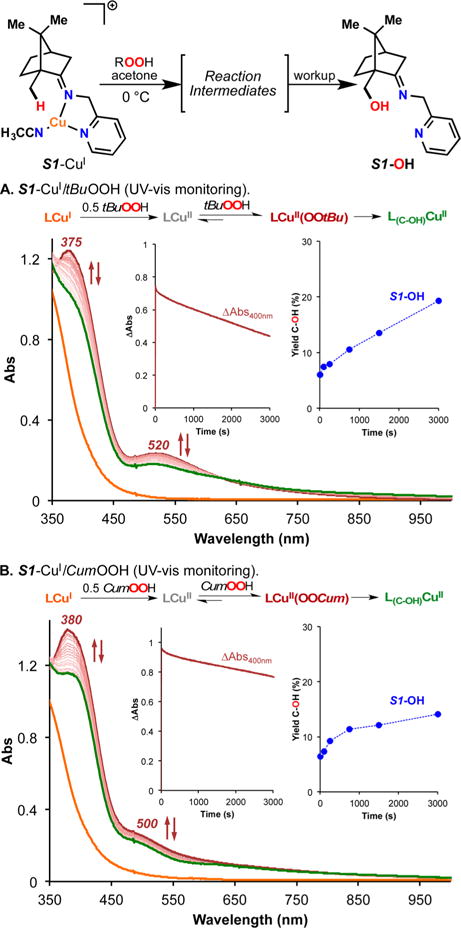
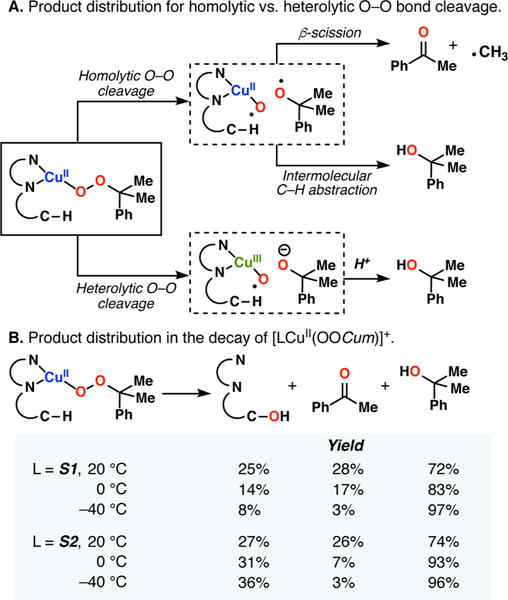
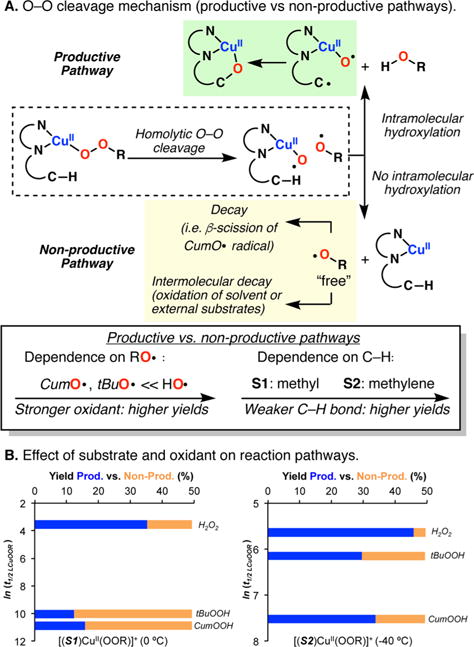
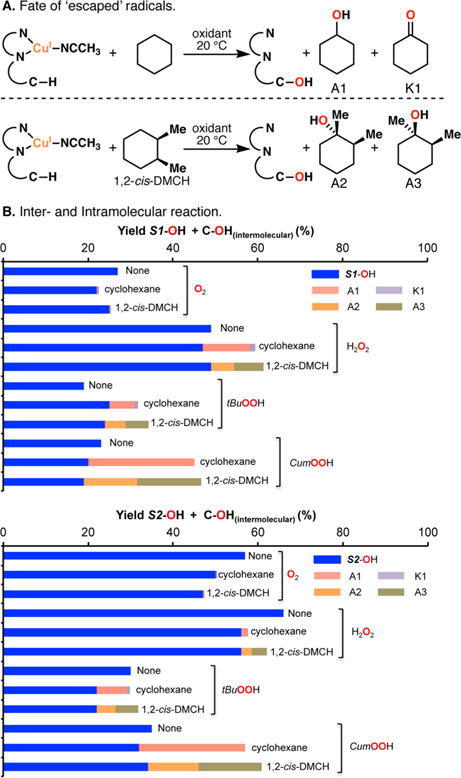
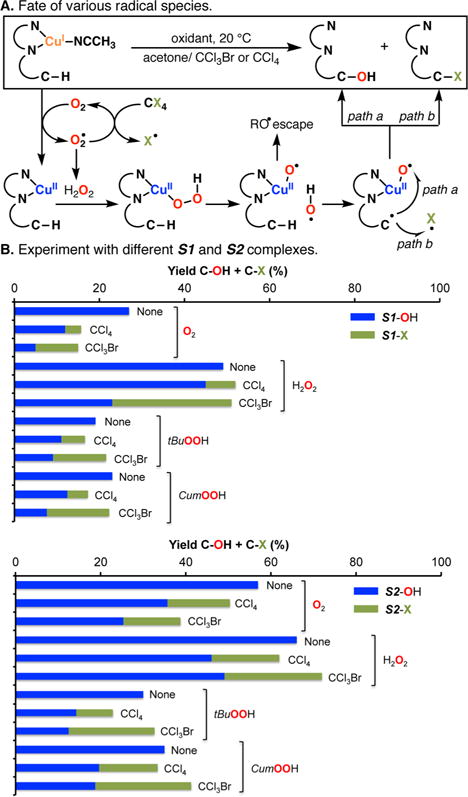
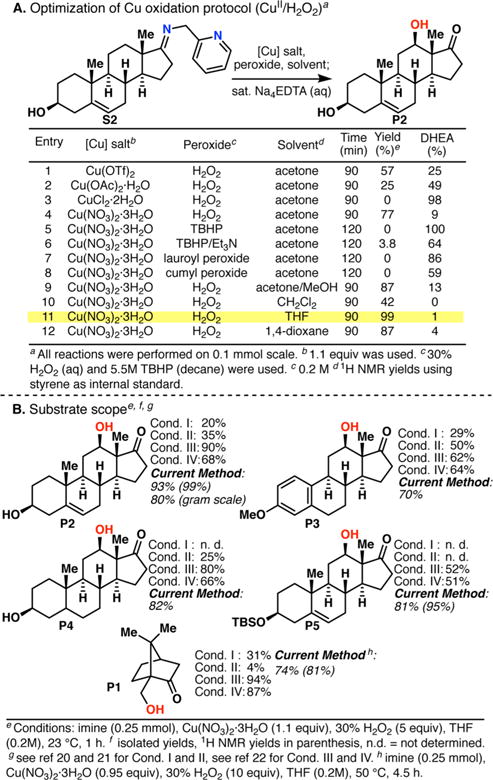
The use of copper in directed C-H oxidation has been relatively underexplored. In a seminal example, Schönecker showed that copper and O2 promoted the hydroxylation of steroid-containing ligands. Recently, Baran (J. Am. Chem. Soc. 2015, 137, 13776) improved the reaction conditions to oxidize similar substrates with excellent yields. In both reports, the involvement of Cu2O2 intermediates was suggested. In this collaborative article, we studied the hydroxylation mechanism in great detail, resulting in the overhaul of the previously accepted mechanism and the development of improved reaction conditions. Extensive experimental evidence (spectroscopic characterization, kinetic analysis, intermolecular reactivity, and radical trap experiments) is provided to support each of the elementary steps proposed and the hypothesis that a key mononuclear LCuII(OOR) intermediate undergoes homolytic O-O cleavage to generate reactive RO• species, which are responsible for key C-H hydroxylation within the solvent cage. These key findings allowed the oxidation protocol to be reformulated, leading to improvements of the reaction cost, practicability, and isolated yield.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
References
-
- Gutekunst WR, Baran PS. Chem Soc Rev. 2011;40:1976. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
