

Critical role of SIK3 in mediating high salt and IL-17 synergy leading to breast cancer cell proliferation
- PMID: 28658303
- PMCID: PMC5489190
- DOI: 10.1371/journal.pone.0180097
Critical role of SIK3 in mediating high salt and IL-17 synergy leading to breast cancer cell proliferation
Abstract
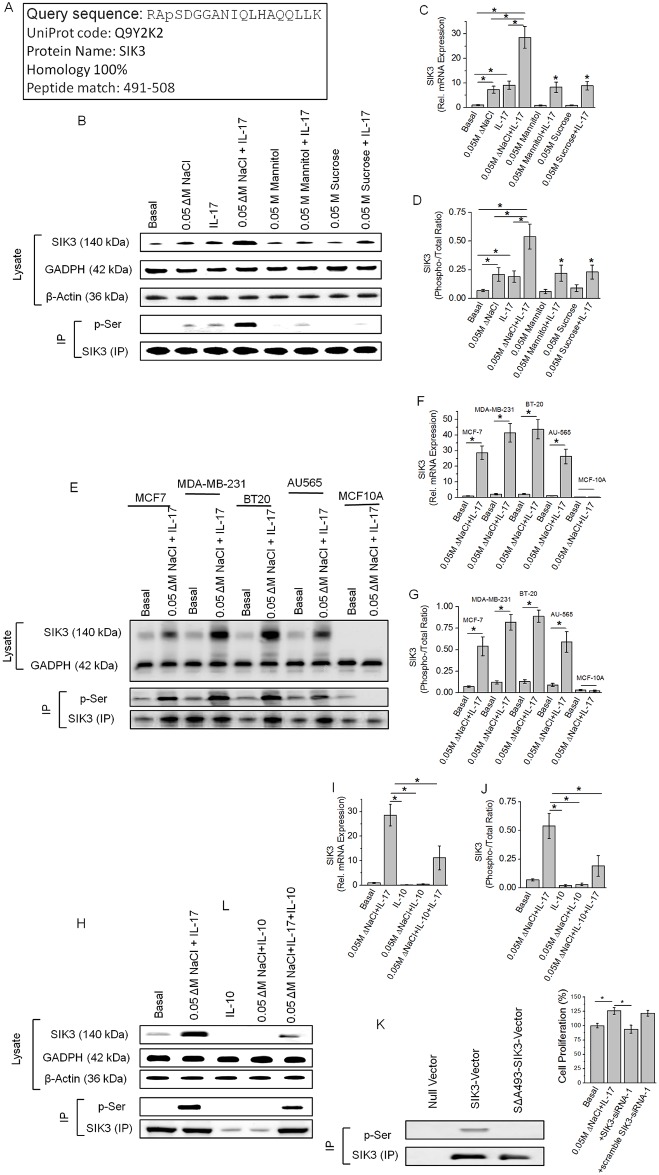
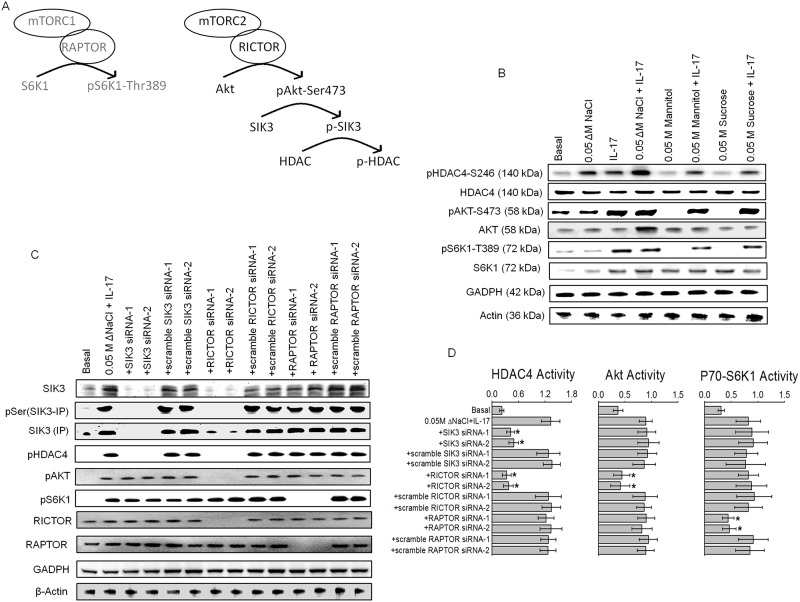
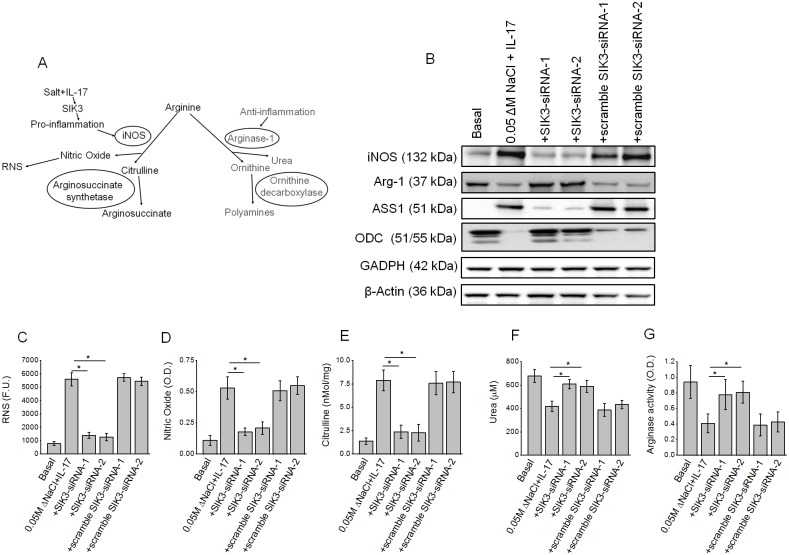
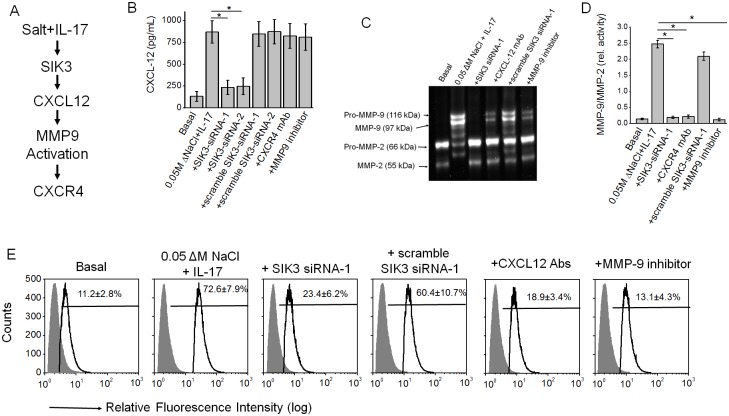
Chronic inflammation is a well-known precursor for cancer development and proliferation. We have recently demonstrated that high salt (NaCl) synergizes with sub-effective interleukin (IL)-17 to induce breast cancer cell proliferation. However, the exact molecular mechanisms mediating this effect are unclear. In our current study, we adopted a phosphoproteomic-based approach to identify salt modulated kinase-proteome specific molecular targets. The phosphoprotemics based binary comparison between heavy labelled MCF-7 cells treated with high salt (Δ0.05 M NaCl) and light labelled MCF-7 cells cultured under basal conditions demonstrated an enhanced phosphorylation of Serine-493 of SIK3 protein. The mRNA transcript and protein expression analysis of SIK3 in MCF-7 cells demonstrated a synergistic enhancement following co-treatment with high salt and sub-effective IL-17 (0.1 ng/mL), as compared to either treatments alone. A similar increase in SIK3 expression was observed in other breast cancer cell lines, MDA-MB-231, BT20, and AU565, while non-malignant breast epithelial cell line, MCF10A, did not induce SIK3 expression under similar conditions. Biochemical studies revealed mTORC2 acted as upstream mediator of SIK3 phosphorylation. Importantly, cell cycle analysis by flow cytometry demonstrated SIK3 induced G0/G1-phase release mediated cell proliferation, while SIK3 silencing abolished this effect. Also, SIK3 induced pro-inflammatory arginine metabolism, as evidenced by upregulation of the enzymes iNOS and ASS-1, along with downregulation of anti-inflammatory enzymes, arginase-1 and ornithine decarboxylase. Furthermore, gelatin zymography analysis has demonstrated that SIK3 induced expression of tumor metastatic CXCR4 through MMP-9 activation. Taken together, our data suggests a critical role of SIK3 in mediating three important hallmarks of cancer namely, cell proliferation, inflammation and metastasis. These studies provide a mechanistic basis for the future utilization of SIK3 as a key drug discovery target to improve breast cancer therapy.
Conflict of interest statement
Figures

References
-
- Crusz SM, Balkwill FR. Inflammation and cancer: advances and new agents. Nat Rev Clin Oncol 2015; 12(10): 584–596. doi: 10.1038/nrclinonc.2015.105 - DOI - PubMed
-
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144(5): 646–674. doi: 10.1016/j.cell.2011.02.013 - DOI - PubMed
-
- Landskron G, De la Fuente M, Thuwajit P, Thuwajit C, Hermoso MA. Chronic inflammation and cytokines in the tumor microenvironment. J Immunol Res 2014; 2014: 149185 doi: 10.1155/2014/149185 - DOI - PMC - PubMed
-
- Bailey SR, Nelson MH, Himes RA, Li Z, Mehrotra S, Paulos CM. Th17 cells in cancer: the ultimate identity crisis. Front Immunol 2014; 5: 276 doi: 10.3389/fimmu.2014.00276 - DOI - PMC - PubMed
-
- Wu C, Yosef N, Thalhamer T, Zhu C, Xiao S, Kishi Y et al. Induction of pathogenic TH17 cells by inducible salt-sensing kinase SGK1. Nature 2013; 496(7446): 513–517. doi: 10.1038/nature11984 - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
