

Amyloid precursor protein and endosomal-lysosomal dysfunction in Alzheimer's disease: inseparable partners in a multifactorial disease
- PMID: 28663518
- PMCID: PMC6137496
- DOI: 10.1096/fj.201700359
Amyloid precursor protein and endosomal-lysosomal dysfunction in Alzheimer's disease: inseparable partners in a multifactorial disease
Abstract
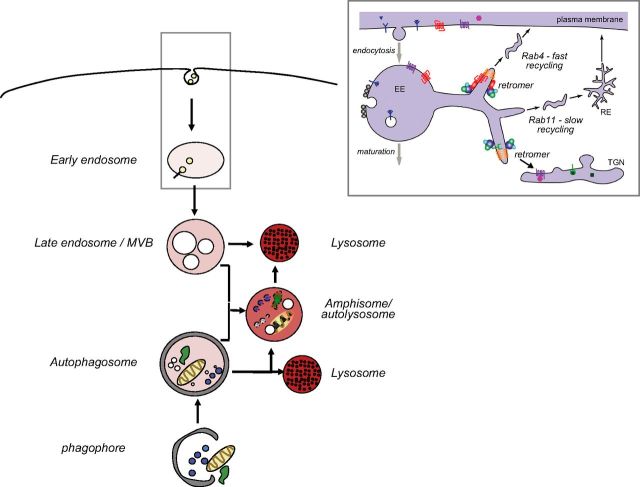
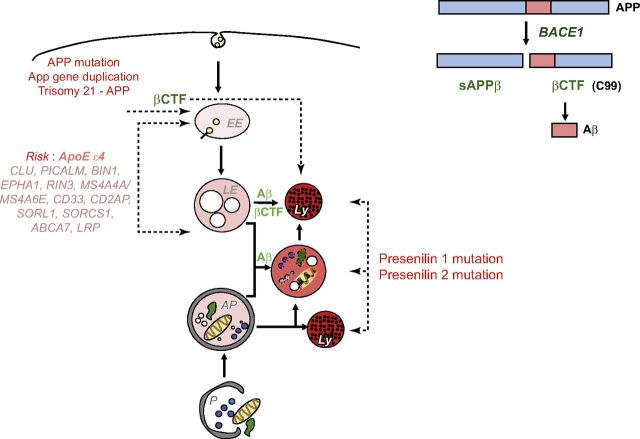
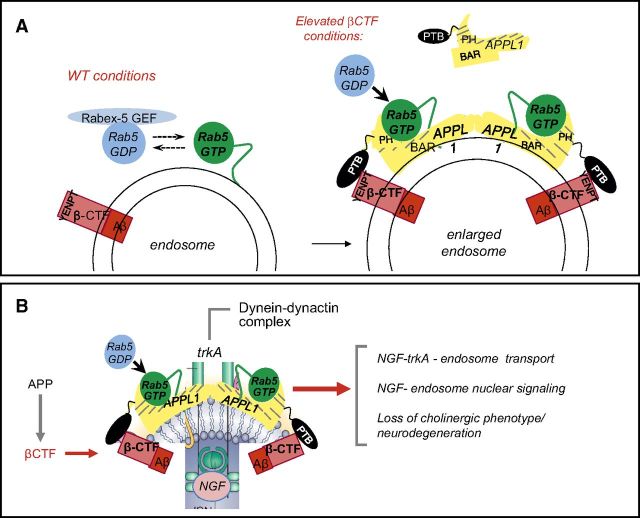
Abnormalities of the endosomal-lysosomal network (ELN) are a signature feature of Alzheimer's disease (AD). These include the earliest known cytopathology that is specific to AD and that affects endosomes and induces the progressive failure of lysosomes, each of which are directly linked by distinct mechanisms to neurodegeneration. The origins of ELN dysfunction and β-amyloidogenesis closely overlap, which reflects their common genetic basis, the established early involvement of endosomes and lysosomes in amyloid precursor protein (APP) processing and clearance, and the pathologic effect of certain APP metabolites on ELN functions. Genes that promote β-amyloidogenesis in AD (APP, PSEN1/2, and APOE4) have primary effects on ELN function. The importance of primary ELN dysfunction to pathogenesis is underscored by the mutations in more than 35 ELN-related genes that, thus far, are known to cause familial neurodegenerative diseases even though different pathogenic proteins may be involved. In this article, I discuss growing evidence that implicates AD gene-driven ELN disruptions as not only the antecedent pathobiology that underlies β-amyloidogenesis but also as the essential partner with APP and its metabolites that drive the development of AD, including tauopathy, synaptic dysfunction, and neurodegeneration. The striking amelioration of diverse deficits in animal AD models by remediating ELN dysfunction further supports a need to integrate APP and ELN relationships, including the role of amyloid-β, into a broader conceptual framework of how AD arises, progresses, and may be effectively therapeutically targeted.-Nixon, R. A. Amyloid precursor protein and endosomal-lysosomal dysfunction in Alzheimer's disease: inseparable partners in a multifactorial disease.
Keywords: Down Syndrome; apoliprotein E; autophagy; cholinergic neurodegeneration; presenilin.
© FASEB.
Conflict of interest statement
Research from R.A.N. laboratories is supported by the U.S. National Institutes of Health, National Institute on Aging. Contributions of Martin Berg (Center for Dementia Research, Nathan S. Kline Institute) to manuscript preparation are gratefully acknowledged.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
