

A Fast Association Test for Identifying Pathogenic Variants Involved in Rare Diseases
- PMID: 28669401
- PMCID: PMC5501869
- DOI: 10.1016/j.ajhg.2017.05.015
A Fast Association Test for Identifying Pathogenic Variants Involved in Rare Diseases
Abstract
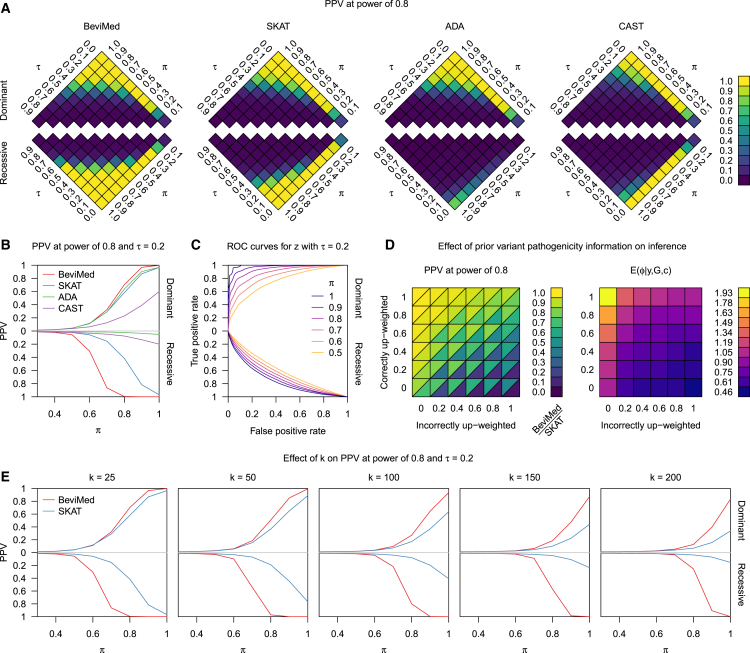
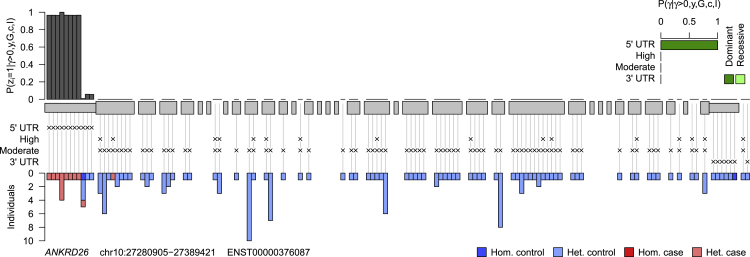
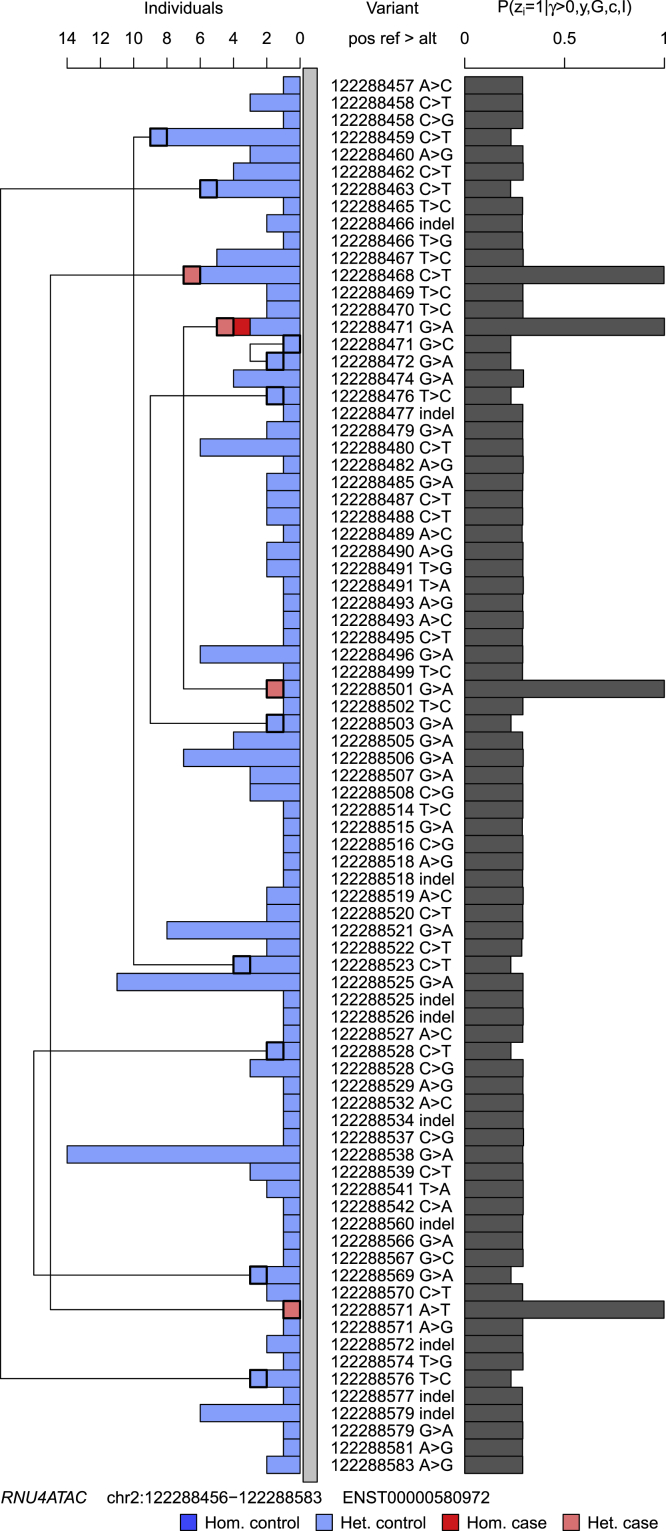
We present a rapid and powerful inference procedure for identifying loci associated with rare hereditary disorders using Bayesian model comparison. Under a baseline model, disease risk is fixed across all individuals in a study. Under an association model, disease risk depends on a latent bipartition of rare variants into pathogenic and non-pathogenic variants, the number of pathogenic alleles that each individual carries, and the mode of inheritance. A parameter indicating presence of an association and the parameters representing the pathogenicity of each variant and the mode of inheritance can be inferred in a Bayesian framework. Variant-specific prior information derived from allele frequency databases, consequence prediction algorithms, or genomic datasets can be integrated into the inference. Association models can be fitted to different subsets of variants in a locus and compared using a model selection procedure. This procedure can improve inference if only a particular class of variants confers disease risk and can suggest particular disease etiologies related to that class. We show that our method, called BeviMed, is more powerful and informative than existing rare variant association methods in the context of dominant and recessive disorders. The high computational efficiency of our algorithm makes it feasible to test for associations in the large non-coding fraction of the genome. We have applied BeviMed to whole-genome sequencing data from 6,586 individuals with diverse rare diseases. We show that it can identify multiple loci involved in rare diseases, while correctly inferring the modes of inheritance, the likely pathogenic variants, and the variant classes responsible.
Keywords: Bayesian inference; Mendelian diseases; hereditary disorders; rare diseases; rare variant association test; rare variants; whole-genome sequencing.
Copyright © 2017 The Author(s). Published by Elsevier Inc. All rights reserved.
Figures
References
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
