

Differential diagnosis of neuromyelitis optica spectrum disorders
- PMID: 28670343
- PMCID: PMC5476332
- DOI: 10.1177/1756285617709723
Differential diagnosis of neuromyelitis optica spectrum disorders
Abstract
Neuromyelitis optica spectrum disorder (NMOSD) is an inflammatory disorder of the central nervous system (CNS) mostly manifesting as optic neuritis and/or myelitis, which are frequently recurrent/bilateral or longitudinally extensive, respectively. As the autoantibody to aquaporin-4 (AQP4-Ab) can mediate the pathogenesis of NMOSD, testing for the AQP4-Ab in serum of patients can play a crucial role in diagnosing NMOSD. Nevertheless, the differential diagnosis of NMOSD in clinical practice is often challenging despite the phenotypical and serological characteristics of the disease because: (1) diverse diseases with autoimmune, vascular, infectious, or neoplastic etiologies can mimic these phenotypes of NMOSD; (2) patients with NMOSD may only have limited clinical manifestations, especially in their early disease stages; (3) test results for AQP4-Ab can be affected by several factors such as assay methods, serologic status, disease stages, or types of treatment; (4) some patients with NMOSD do not have AQP4-Ab; and (5) test results for the AQP4-Ab may not be readily available for the acute management of patients. Despite some similarity in their phenotypes, these NMOSD and NMOSD-mimics are distinct from each other in their pathogenesis, prognosis, and most importantly treatment. Understanding the detailed clinical, serological, radiological, and prognostic differences of these diseases will improve the proper management as well as diagnosis of patients.
Keywords: Devic’s disease; aquaporin-4 antibody; differential diagnosis; longitudinally extensive transverse myelitis; multiple sclerosis; myelin oligodendrocyte glycoprotein antibody; neuromyelitis optica spectrum disorders; optic neuritis.
Conflict of interest statement
Conflict of interest statement: Prof. Kazuo Fujihara serves on scientific advisory boards for Bayer Schering Pharma, Biogen Idec, Mitsubishi Tanabe Pharma Corporation, Novartis Pharma, Chugai Pharmaceutical, Ono Pharmaceutical, Nihon Pharmaceutical, Merck Serono, Alexion Pharmaceuticals, Medimmune and Medical Review; has received funding for travel and speaker honoraria from Bayer Schering Pharma, Biogen Idec, Eisai Inc., Mitsubishi Tanabe Pharma Corporation, Novartis Pharma, Astellas Pharma Inc., Takeda Pharmaceutical Company Limited, Asahi Kasei Medical Co., Daiichi Sankyo, and Nihon Pharmaceutical; serves as an editorial board member of Clinical and Experimental Neuroimmunology (2009 to present) and an advisory board member of the Sri Lanka Journal of Neurology; has received research support from Bayer Schering Pharma, Biogen Idec Japan, Asahi Kasei Medical, The Chemo-Sero-Therapeutic Research Institute, Teva Pharmaceutical, Mitsubishi Tanabe Pharma, Teijin Pharma, Chugai Pharmaceutical, Ono Pharmaceutical, Nihon Pharmaceutical, and Genzyme Japan.
Figures
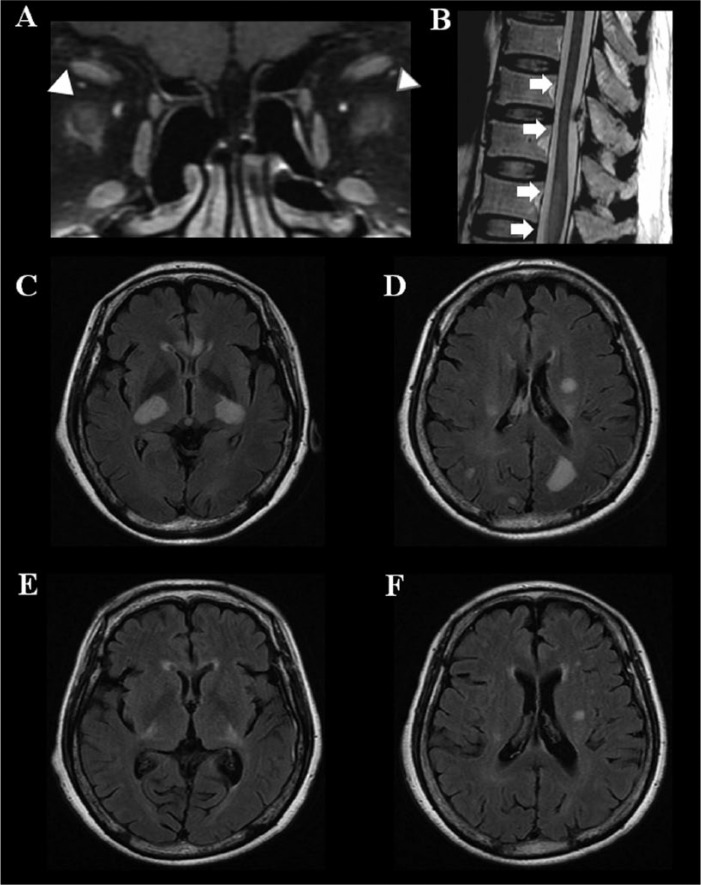
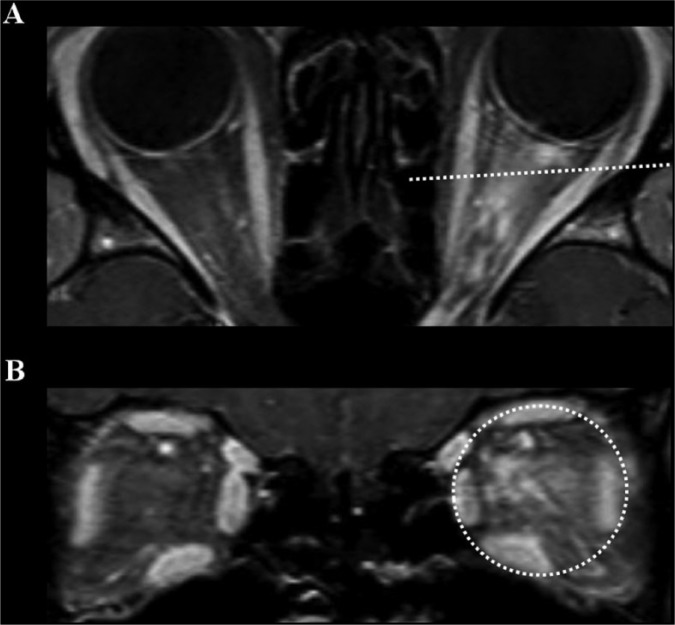

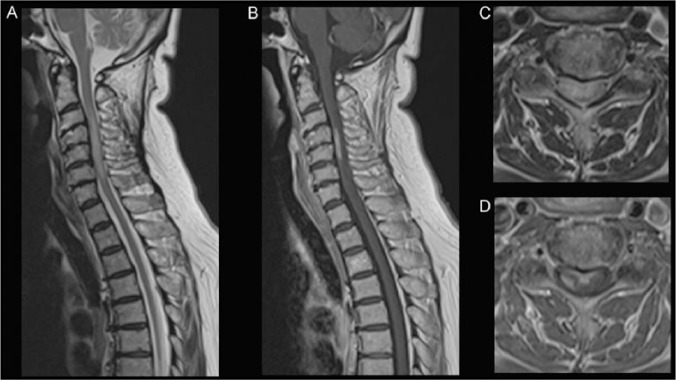
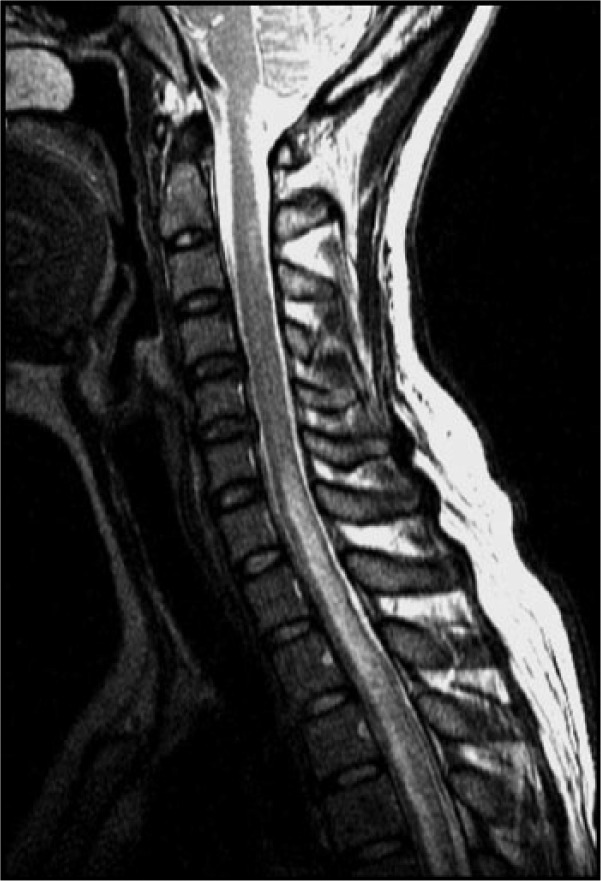
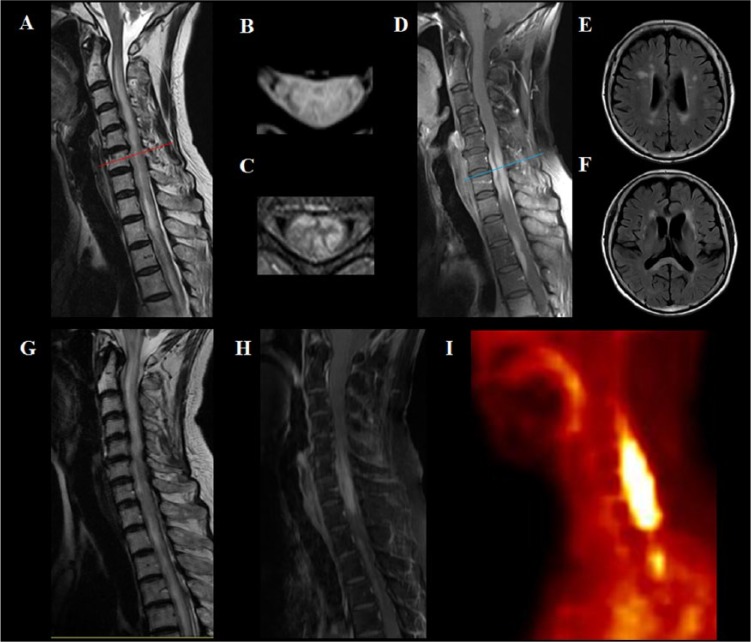
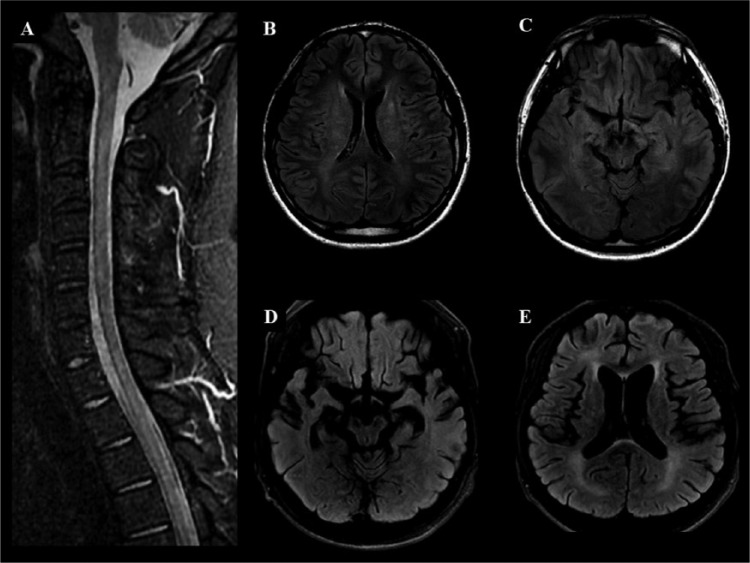
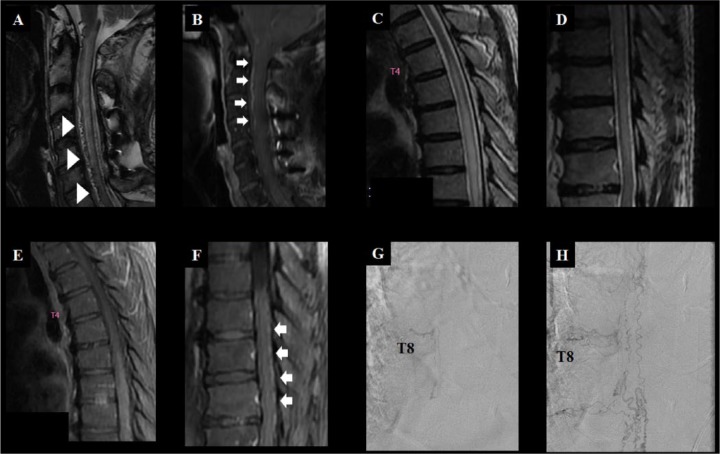
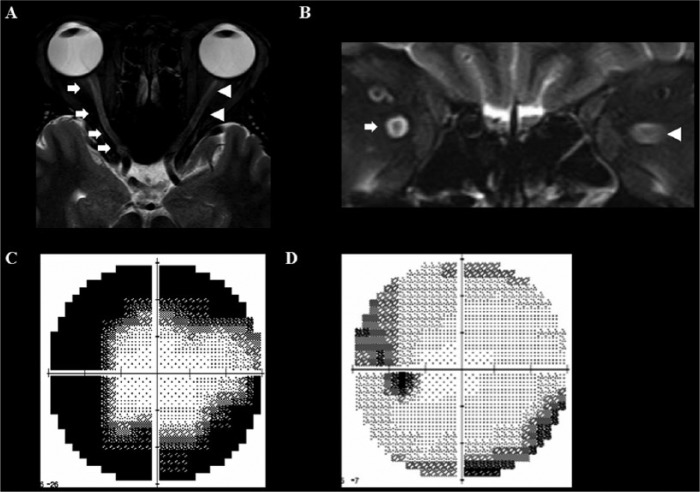
References
-
- Wingerchuk D. Neuromyelitis optica: effect of gender. J Neurol Sci 2009; 286: 18–23. - PubMed
-
- Kitley J, Leite M, Nakashima I, et al. Prognostic factors and disease course in aquaporin-4 antibody-positive patients with neuromyelitis optica spectrum disorder from the United Kingdom and Japan. Brain 2012; 135: 1834–1849. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
