

LGI1 tunes intrinsic excitability by regulating the density of axonal Kv1 channels
- PMID: 28673977
- PMCID: PMC5530646
- DOI: 10.1073/pnas.1618656114
LGI1 tunes intrinsic excitability by regulating the density of axonal Kv1 channels
Abstract
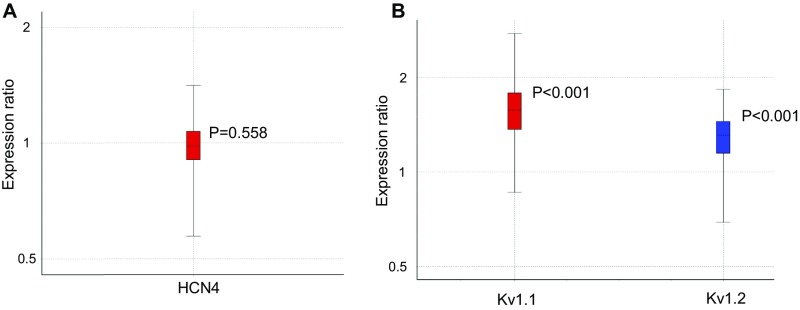
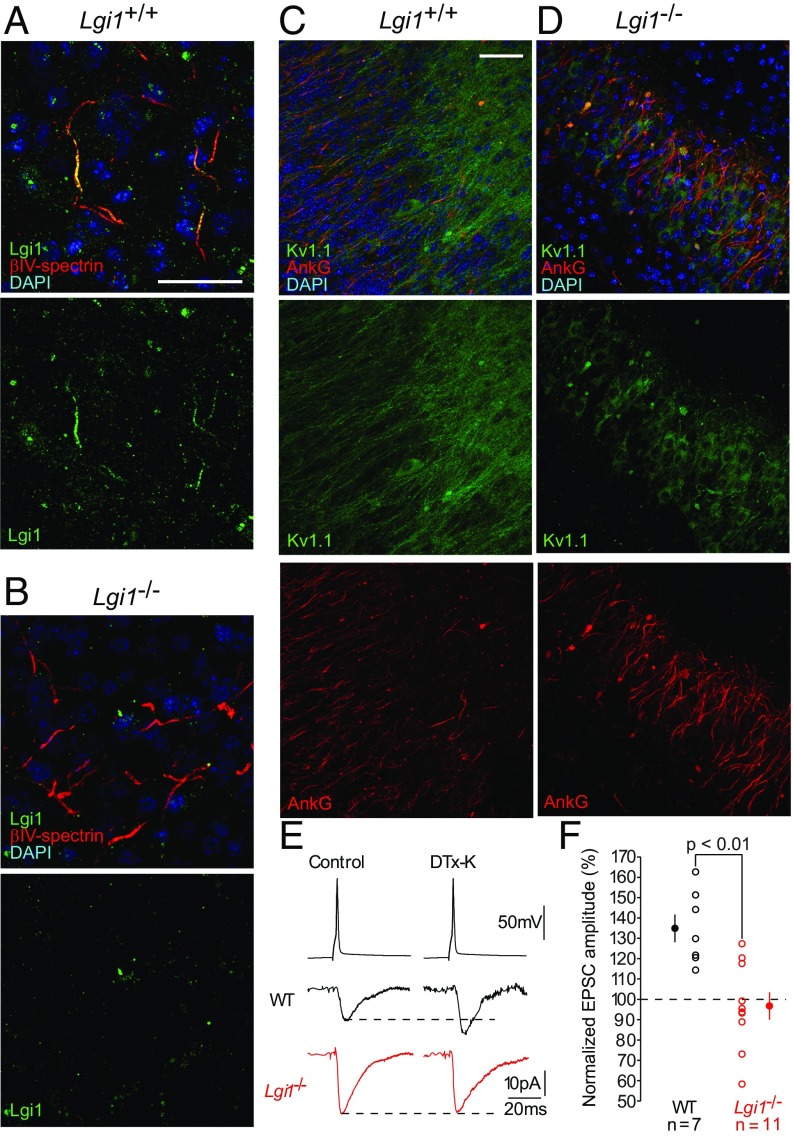
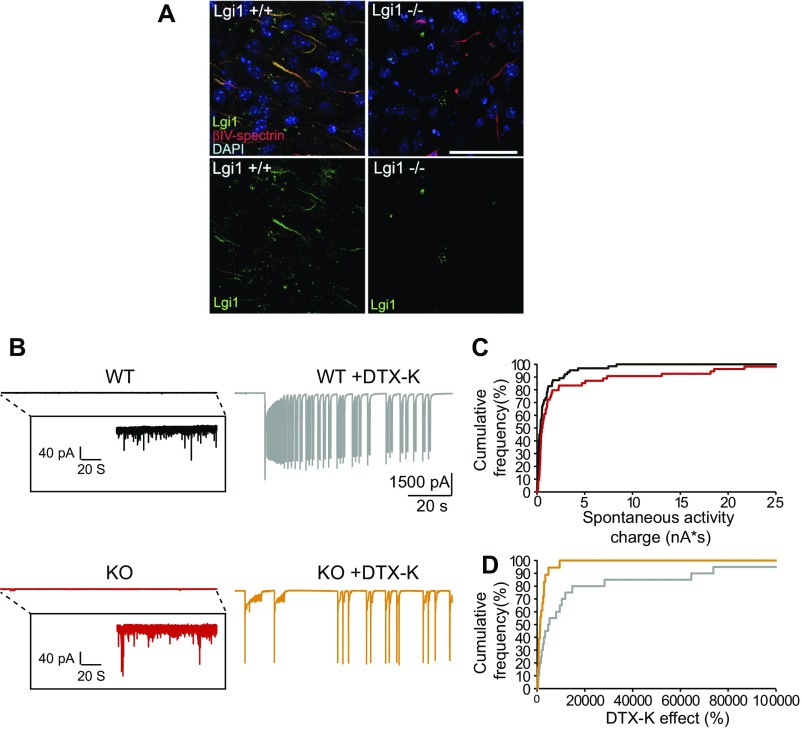
Autosomal dominant epilepsy with auditory features results from mutations in leucine-rich glioma-inactivated 1 (LGI1), a soluble glycoprotein secreted by neurons. Animal models of LGI1 depletion display spontaneous seizures, however, the function of LGI1 and the mechanisms by which deficiency leads to epilepsy are unknown. We investigated the effects of pure recombinant LGI1 and genetic depletion on intrinsic excitability, in the absence of synaptic input, in hippocampal CA3 neurons, a classical focus for epileptogenesis. Our data indicate that LGI1 is expressed at the axonal initial segment and regulates action potential firing by setting the density of the axonal Kv1.1 channels that underlie dendrotoxin-sensitive D-type potassium current. LGI1 deficiency incurs a >50% down-regulation of the expression of Kv1.1 and Kv1.2 via a posttranscriptional mechanism, resulting in a reduction in the capacity of axonal D-type current to limit glutamate release, thus contributing to epileptogenesis.
Keywords: D-type current; Kv1 channels; LGI1; epilepsy; intrinsic excitability.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
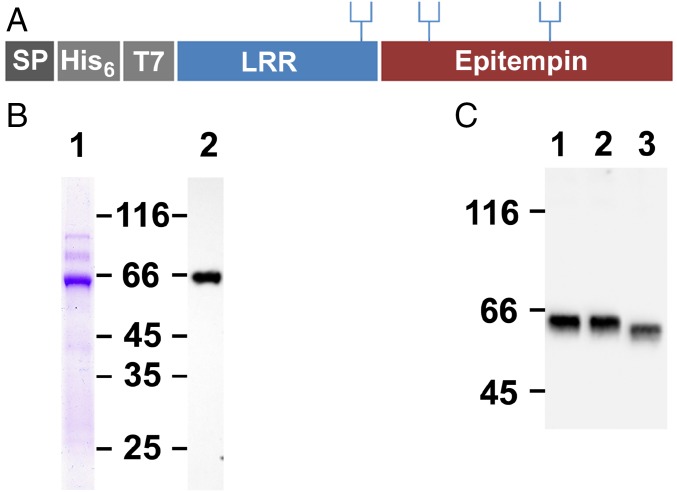
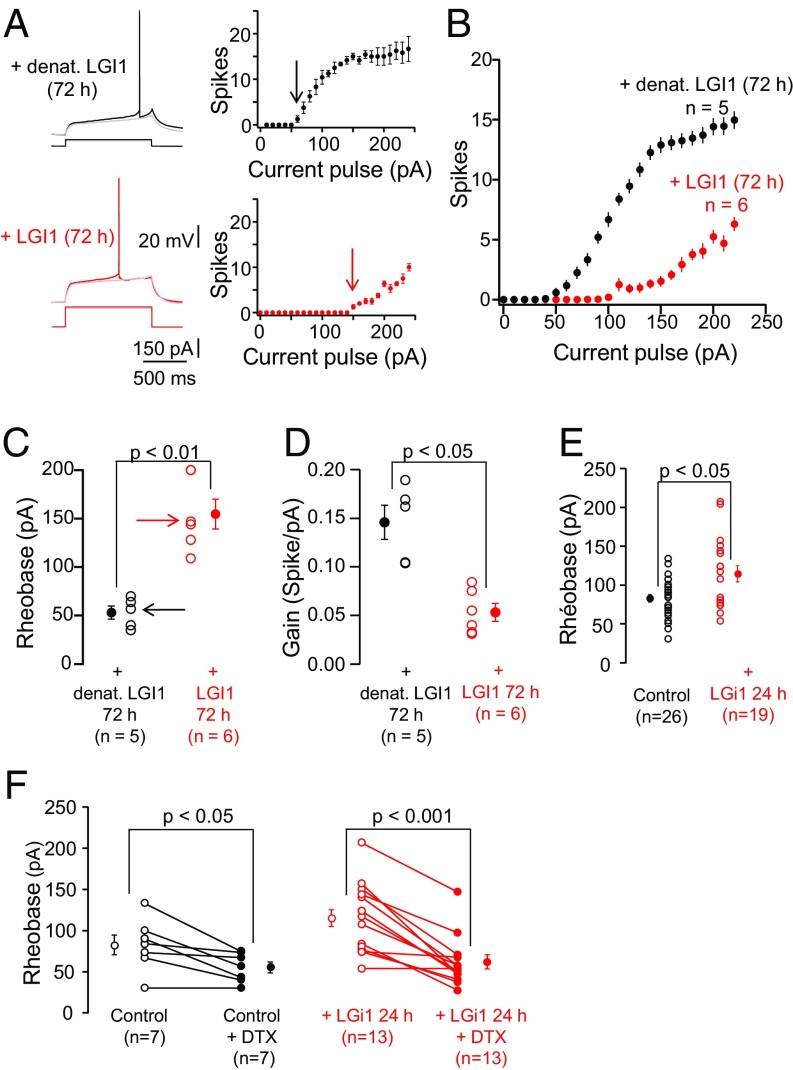
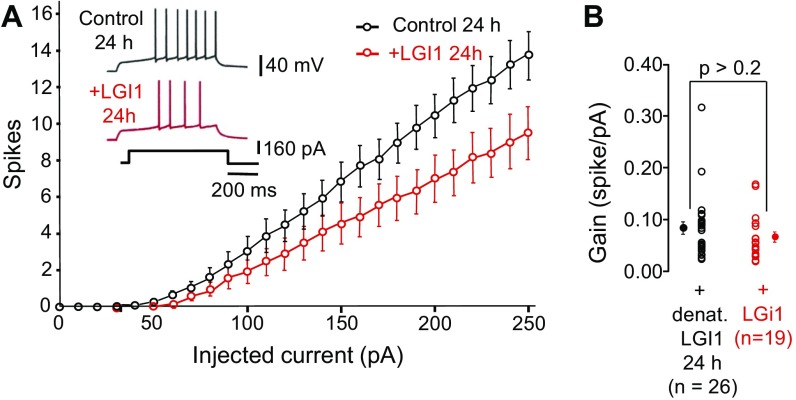
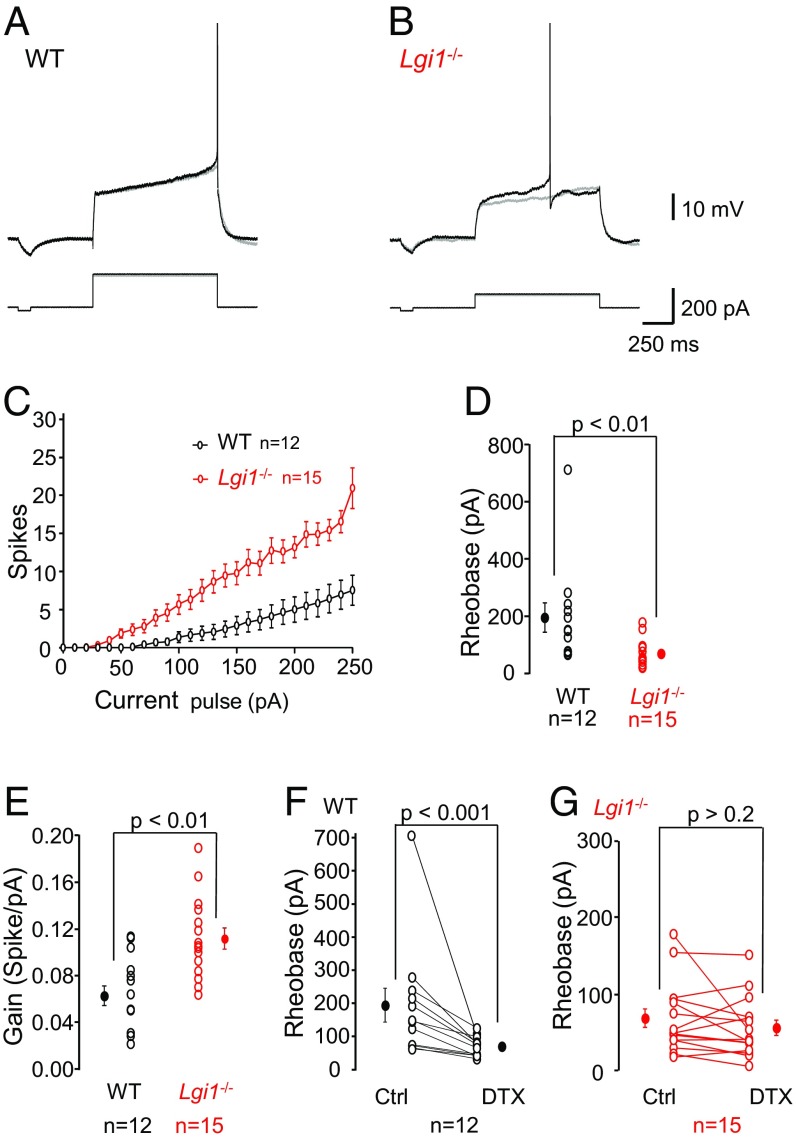
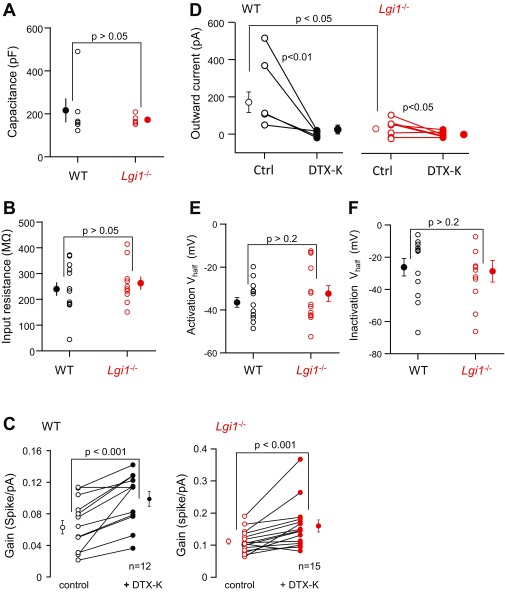
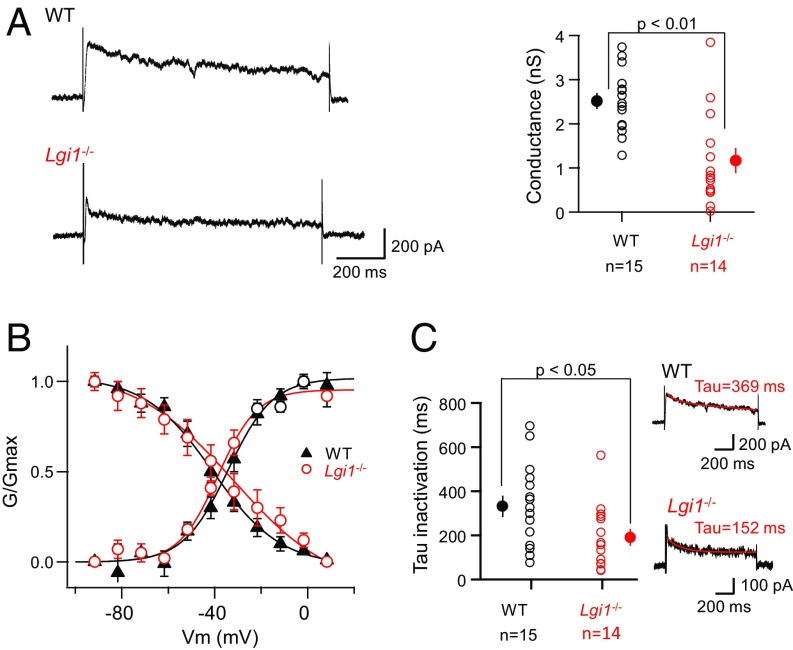
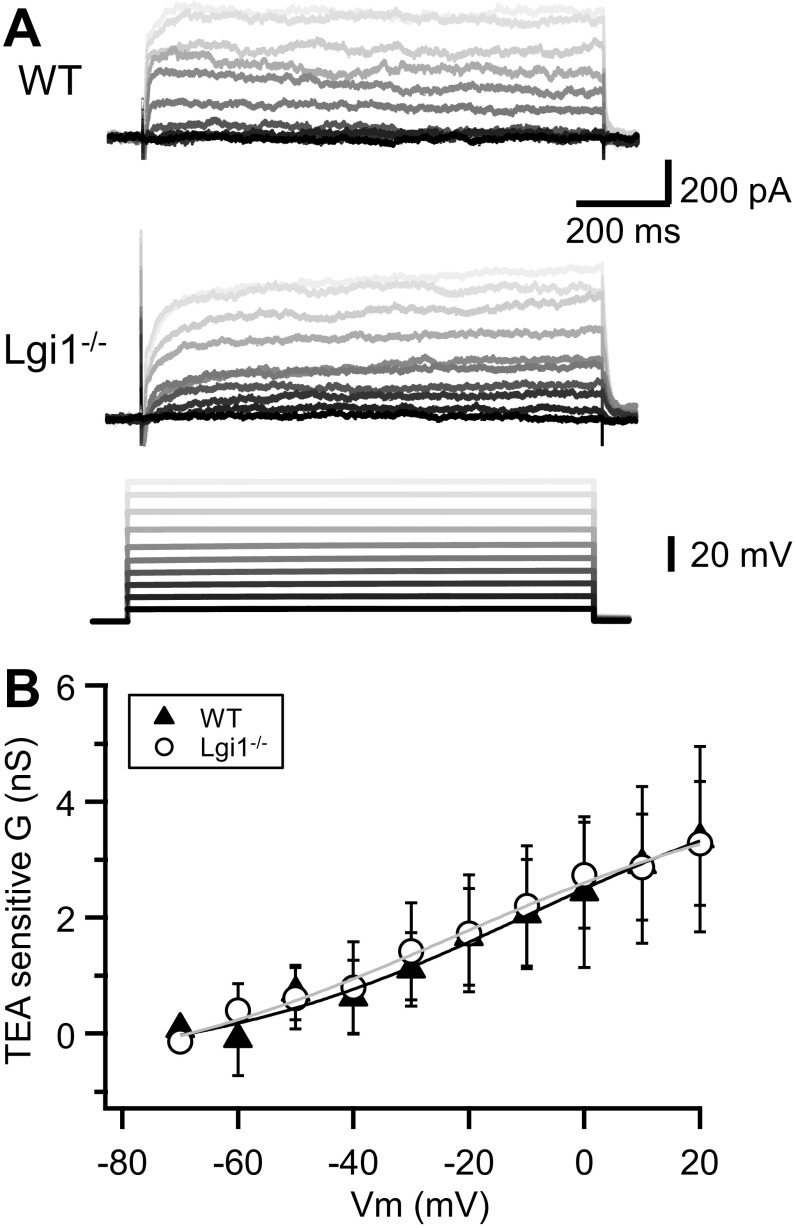
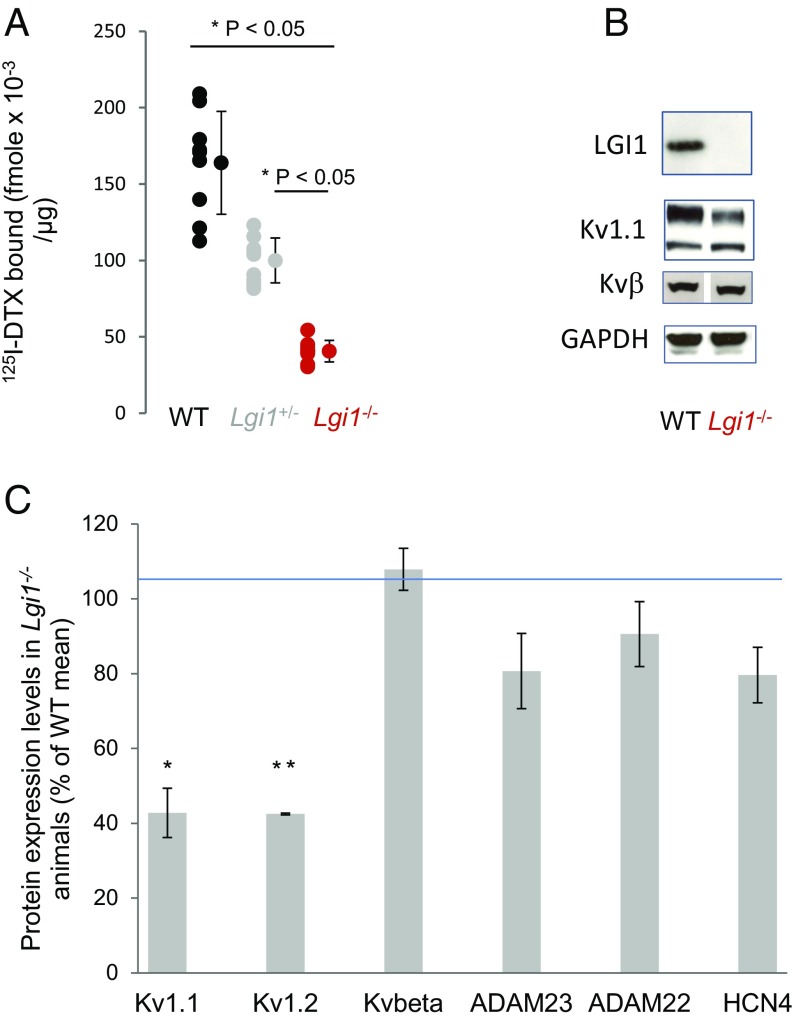
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
