

Quantitative Protein Topography Measurements by High Resolution Hydroxyl Radical Protein Footprinting Enable Accurate Molecular Model Selection
- PMID: 28674401
- PMCID: PMC5495787
- DOI: 10.1038/s41598-017-04689-3
Quantitative Protein Topography Measurements by High Resolution Hydroxyl Radical Protein Footprinting Enable Accurate Molecular Model Selection
Abstract
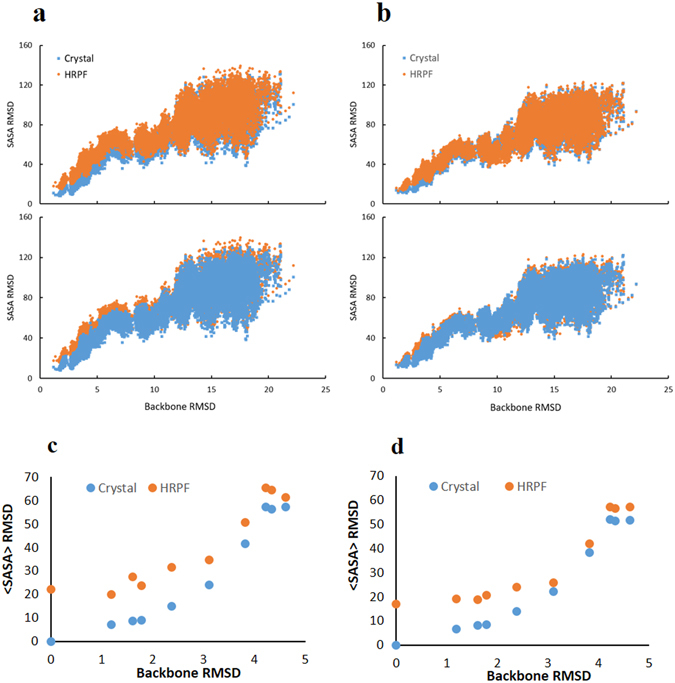
We report an integrated workflow that allows mass spectrometry-based high-resolution hydroxyl radical protein footprinting (HR-HRPF) measurements to accurately measure the absolute average solvent accessible surface area (<SASA>) of amino acid side chains. This approach is based on application of multi-point HR-HRPF, electron-transfer dissociation (ETD) tandem MS (MS/MS) acquisition, measurement of effective radical doses by radical dosimetry, and proper normalization of the inherent reactivity of the amino acids. The accuracy of the resulting <SASA> measurements was tested by using well-characterized protein models. Moreover, we demonstrated the ability to use <SASA> measurements from HR-HRPF to differentiate molecular models of high accuracy (<3 Å backbone RMSD) from models of lower accuracy (>4 Å backbone RMSD). The ability of <SASA> data from HR-HRPF to differentiate molecular model quality was found to be comparable to that of <SASA> data obtained from X-ray crystal structures, indicating the accuracy and utility of HR-HRPF for evaluating the accuracy of computational models.
Conflict of interest statement
J.S.S. discloses a significant ownership share of Photochem Technologies, LLC, a small company that is active in the area of hydroxyl radical protein footprinting instrument design.
Figures




References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
