

HDAC6: A Novel Histone Deacetylase Implicated in Pulmonary Arterial Hypertension
- PMID: 28674407
- PMCID: PMC5495763
- DOI: 10.1038/s41598-017-04874-4
HDAC6: A Novel Histone Deacetylase Implicated in Pulmonary Arterial Hypertension
Abstract
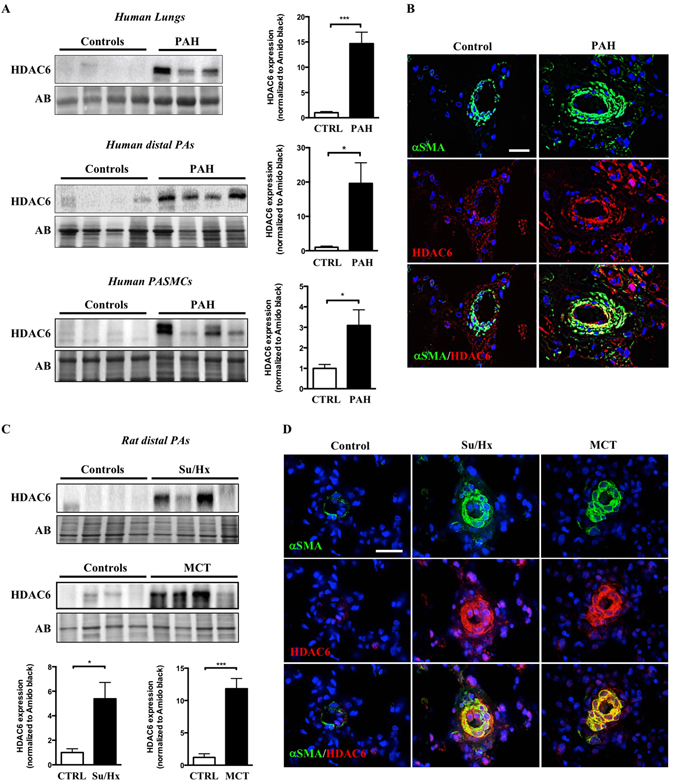
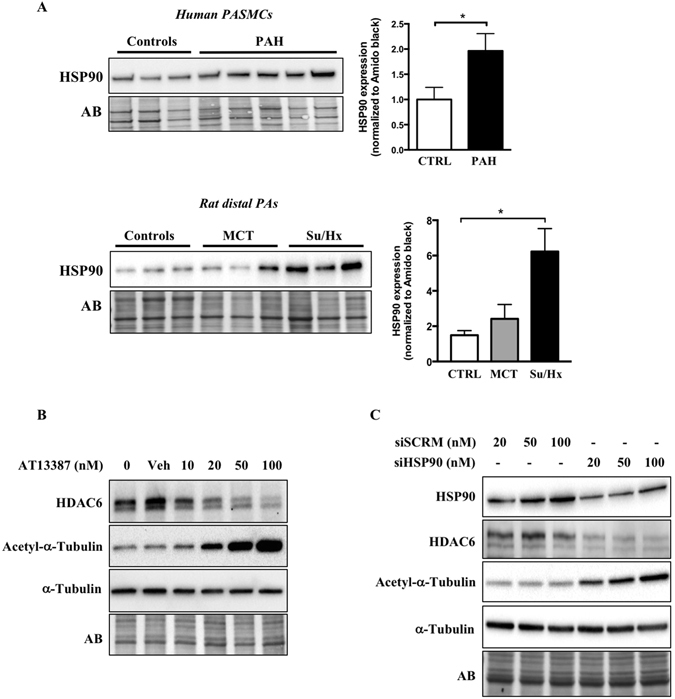
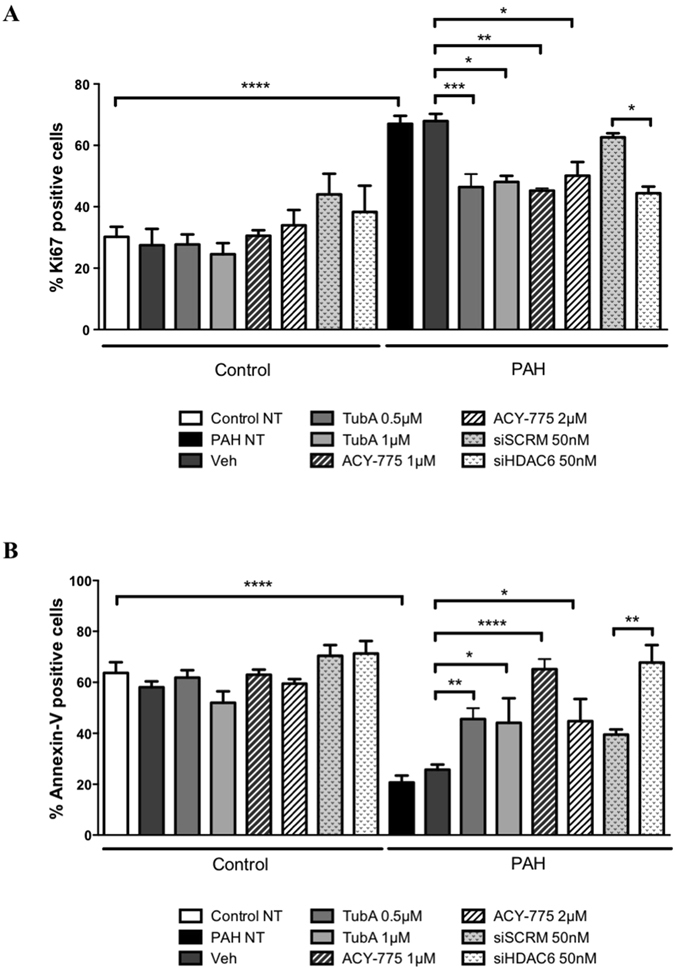
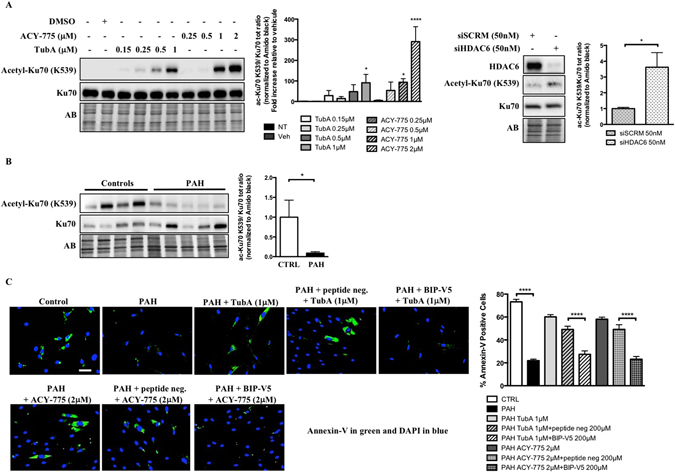
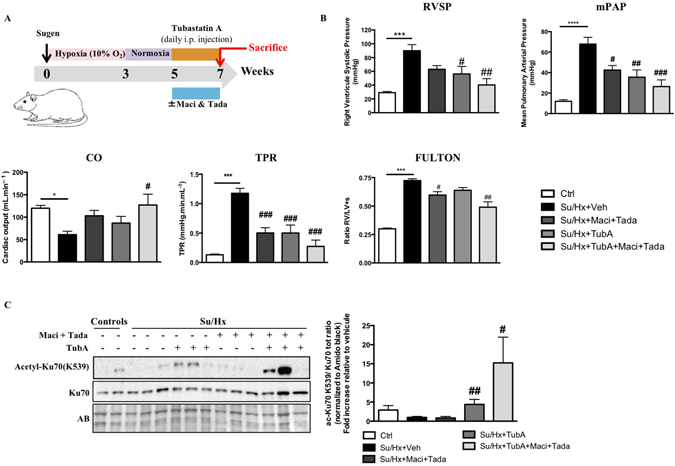
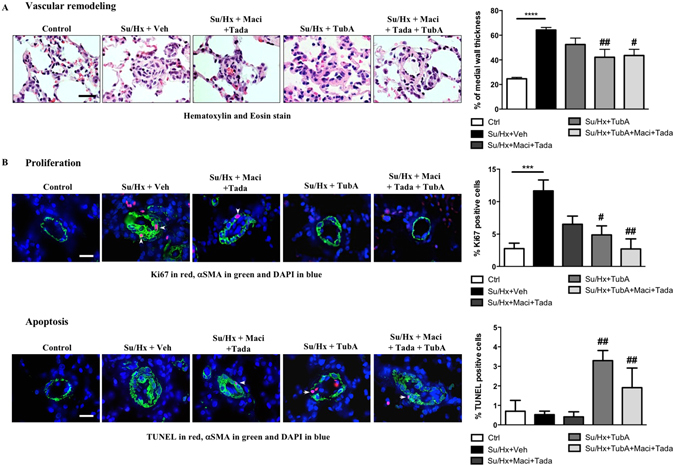
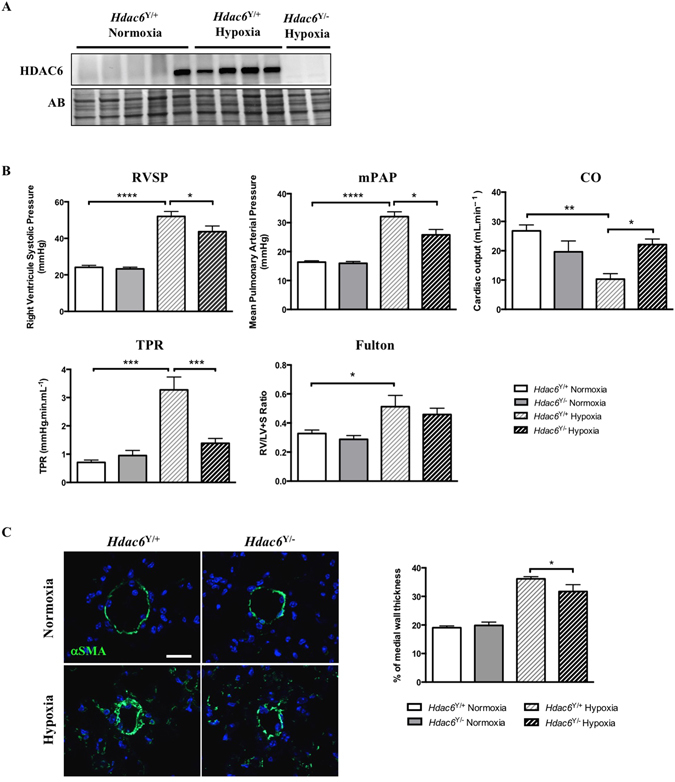
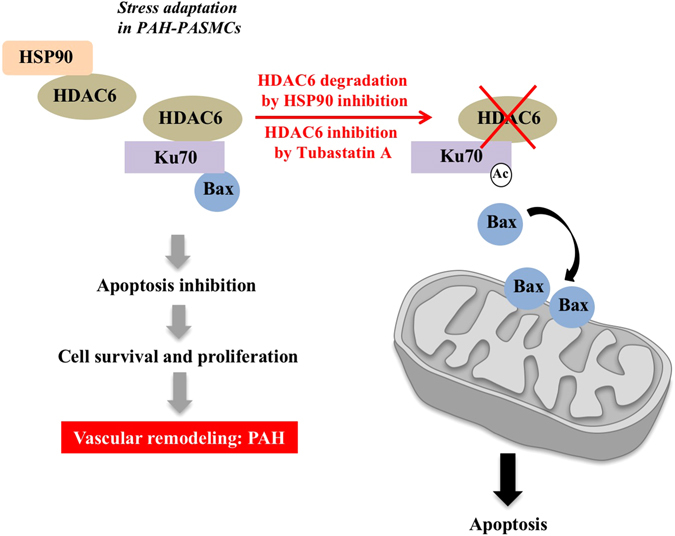
Pulmonary arterial hypertension (PAH) is a vascular remodeling disease with limited therapeutic options. Although exposed to stressful conditions, pulmonary artery (PA) smooth muscle cells (PASMCs) exhibit a "cancer-like" pro-proliferative and anti-apoptotic phenotype. HDAC6 is a cytoplasmic histone deacetylase regulating multiple pro-survival mechanisms and overexpressed in response to stress in cancer cells. Due to the similarities between cancer and PAH, we hypothesized that HDAC6 expression is increased in PAH-PASMCs to face stress allowing them to survive and proliferate, thus contributing to vascular remodeling in PAH. We found that HDAC6 is significantly up-regulated in lungs, distal PAs, and isolated PASMCs from PAH patients and animal models. Inhibition of HDAC6 reduced PAH-PASMC proliferation and resistance to apoptosis in vitro sparing control cells. Mechanistically, we demonstrated that HDAC6 maintains Ku70 in a hypoacetylated state, blocking the translocation of Bax to mitochondria and preventing apoptosis. In vivo, pharmacological inhibition of HDAC6 improved established PAH in two experimental models and can be safely given in combination with currently approved PAH therapies. Moreover, Hdac6 deficient mice were partially protected against chronic hypoxia-induced pulmonary hypertension. Our study shows for the first time that HDAC6 is implicated in PAH development and represents a new promising target to improve PAH.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
