

AAA-ATPases in Protein Degradation
- PMID: 28676851
- PMCID: PMC5476697
- DOI: 10.3389/fmolb.2017.00042
AAA-ATPases in Protein Degradation
Abstract
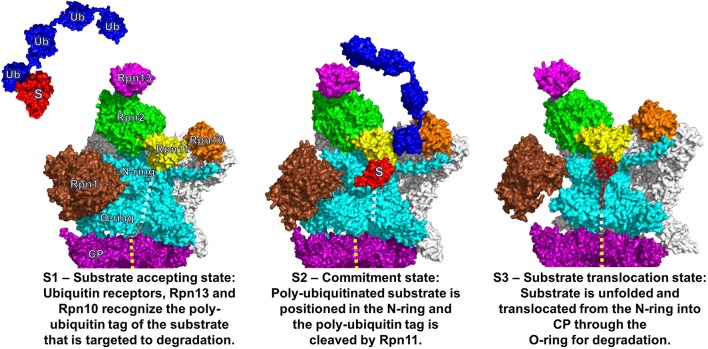
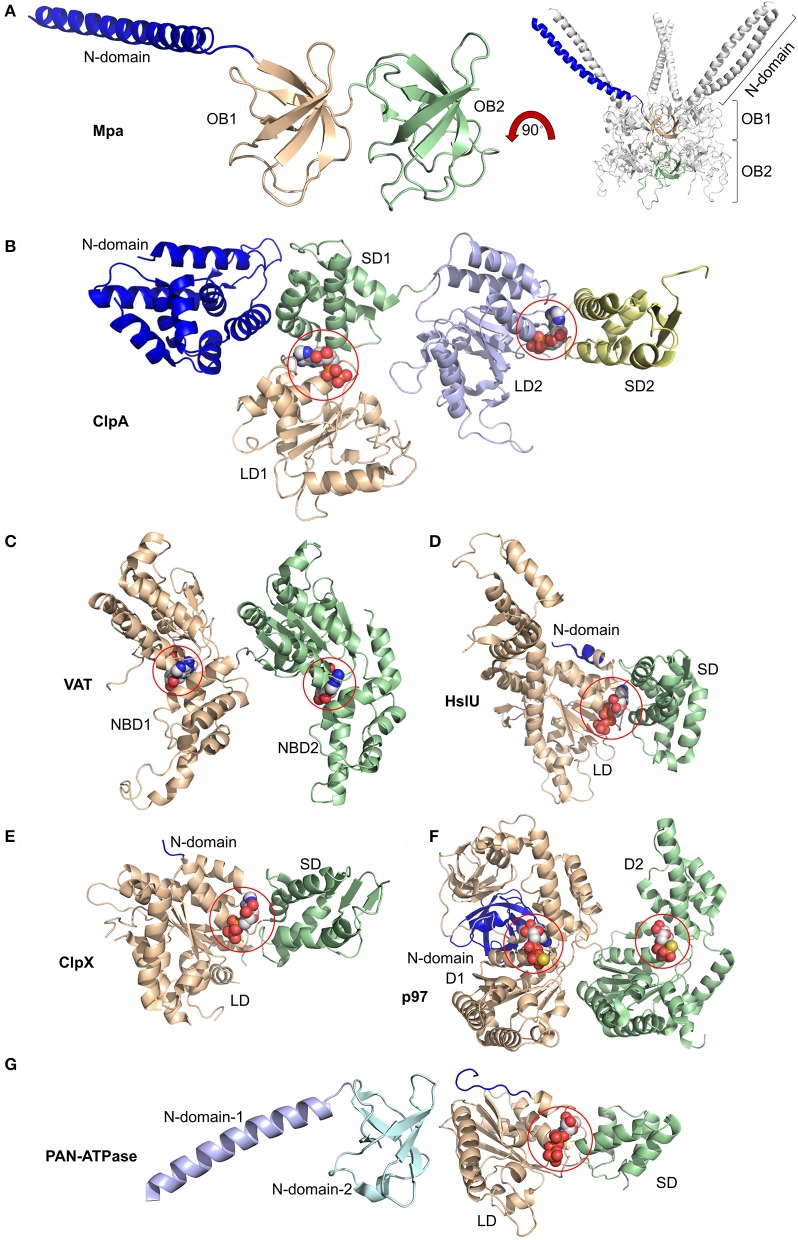
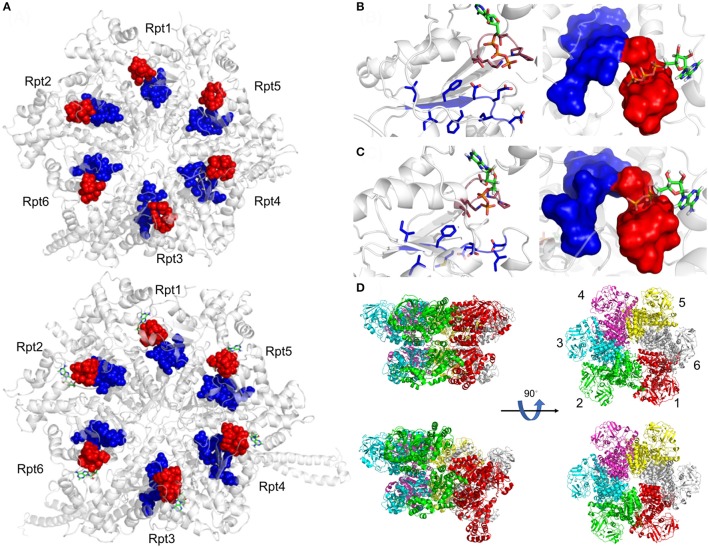
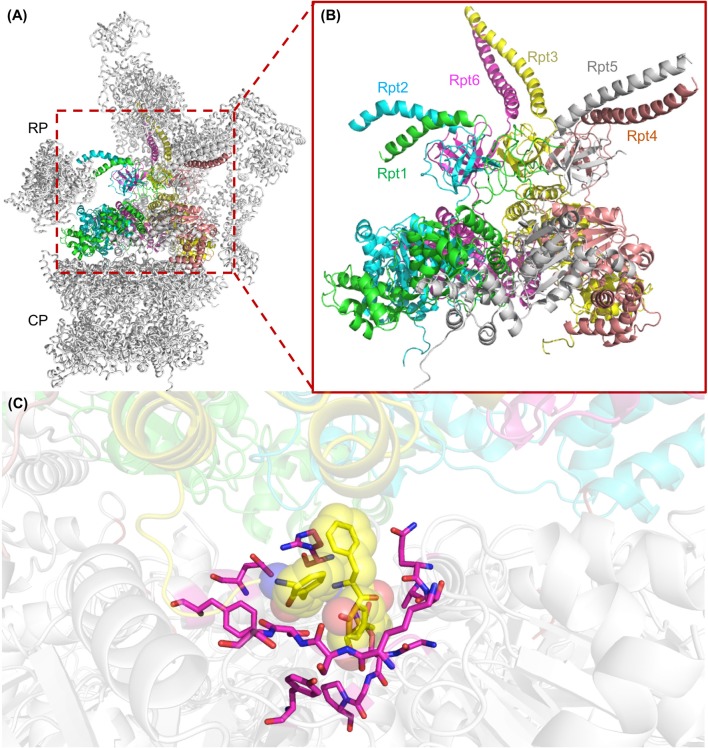
Proteolytic machineries containing multisubunit protease complexes and AAA-ATPases play a key role in protein quality control and the regulation of protein homeostasis. In these protein degradation machineries, the proteolytically active sites are formed by either threonines or serines which are buried inside interior cavities of cylinder-shaped complexes. In eukaryotic cells, the proteasome is the most prominent protease complex harboring AAA-ATPases. To degrade protein substrates, the gates of the axial entry ports of the protease need to be open. Gate opening is accomplished by AAA-ATPases, which form a hexameric ring flanking the entry ports of the protease. Protein substrates with unstructured domains can loop into the entry ports without the assistance of AAA-ATPases. However, folded proteins require the action of AAA-ATPases to unveil an unstructured terminus or domain. Cycles of ATP binding/hydrolysis fuel the unfolding of protein substrates which are gripped by loops lining up the central pore of the AAA-ATPase ring. The AAA-ATPases pull on the unfolded polypeptide chain for translocation into the proteolytic cavity of the protease. Conformational changes within the AAA-ATPase ring and the adjacent protease chamber create a peristaltic movement for substrate degradation. The review focuses on new technologies toward the understanding of the function and structure of AAA-ATPases to achieve substrate recognition, unfolding and translocation into proteasomes in yeast and mammalian cells and into proteasome-equivalent proteases in bacteria and archaea.
Keywords: AAA; ATPase; proteasome; protein folding; proteolysis.
Figures
Similar articles
-
AAA+ proteases: ATP-fueled machines of protein destruction.Annu Rev Biochem. 2011;80:587-612. doi: 10.1146/annurev-biochem-060408-172623. Annu Rev Biochem. 2011. PMID: 21469952 Review.
-
AAA+ ATPases in Protein Degradation: Structures, Functions and Mechanisms.Biomolecules. 2020 Apr 18;10(4):629. doi: 10.3390/biom10040629. Biomolecules. 2020. PMID: 32325699 Free PMC article. Review.
-
Proteasomal AAA-ATPases: structure and function.Biochim Biophys Acta. 2012 Jan;1823(1):67-82. doi: 10.1016/j.bbamcr.2011.07.009. Epub 2011 Jul 23. Biochim Biophys Acta. 2012. PMID: 21820014 Review.
-
Structural alteration in the pore motif of the bacterial 20S proteasome homolog HslV leads to uncontrolled protein degradation.J Mol Biol. 2013 Aug 23;425(16):2940-54. doi: 10.1016/j.jmb.2013.05.011. Epub 2013 May 21. J Mol Biol. 2013. PMID: 23707406
-
Proteasomes and their associated ATPases: a destructive combination.J Struct Biol. 2006 Oct;156(1):72-83. doi: 10.1016/j.jsb.2006.04.012. Epub 2006 May 8. J Struct Biol. 2006. PMID: 16919475 Review.
Cited by
-
Genetic Alterations in Members of the Proteasome 26S Subunit, AAA-ATPase (PSMC) Gene Family in the Light of Proteasome Inhibitor Resistance in Multiple Myeloma.Cancers (Basel). 2023 Jan 15;15(2):532. doi: 10.3390/cancers15020532. Cancers (Basel). 2023. PMID: 36672481 Free PMC article.
-
Natural immunity stimulation using ELICE16INDURES® plant conditioner in field culture of soybean.Heliyon. 2023 Jan 10;9(1):e12907. doi: 10.1016/j.heliyon.2023.e12907. eCollection 2023 Jan. Heliyon. 2023. PMID: 36691550 Free PMC article.
-
Targeted Protein Degradation: Principles and Applications of the Proteasome.Cells. 2023 Jul 13;12(14):1846. doi: 10.3390/cells12141846. Cells. 2023. PMID: 37508510 Free PMC article. Review.
-
GbCYP86A1-1 from Gossypium barbadense positively regulates defence against Verticillium dahliae by cell wall modification and activation of immune pathways.Plant Biotechnol J. 2020 Jan;18(1):222-238. doi: 10.1111/pbi.13190. Epub 2019 Jun 26. Plant Biotechnol J. 2020. PMID: 31207065 Free PMC article.
-
Proteasome inhibition as a therapeutic target for the fungal pathogen Cryptococcus neoformans.Microbiol Spectr. 2023 Sep 26;11(5):e0190423. doi: 10.1128/spectrum.01904-23. Online ahead of print. Microbiol Spectr. 2023. PMID: 37750732 Free PMC article.
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources