

Increasing quality, throughput and speed of sample preparation for strand-specific messenger RNA sequencing
- PMID: 28679365
- PMCID: PMC5499059
- DOI: 10.1186/s12864-017-3900-6
Increasing quality, throughput and speed of sample preparation for strand-specific messenger RNA sequencing
Abstract
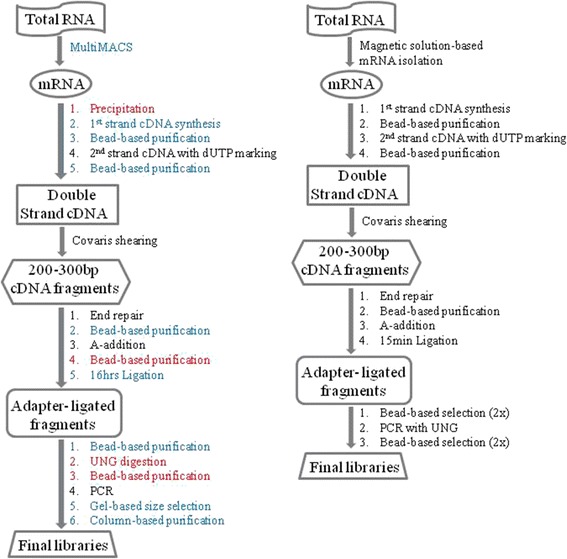
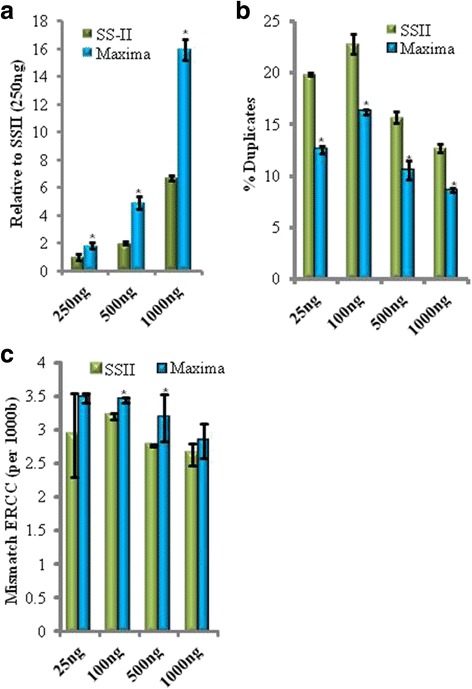
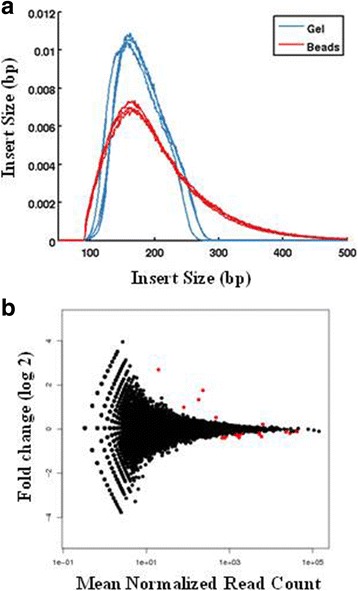
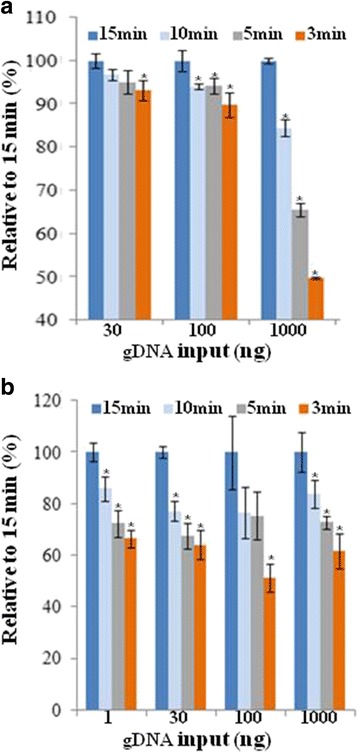
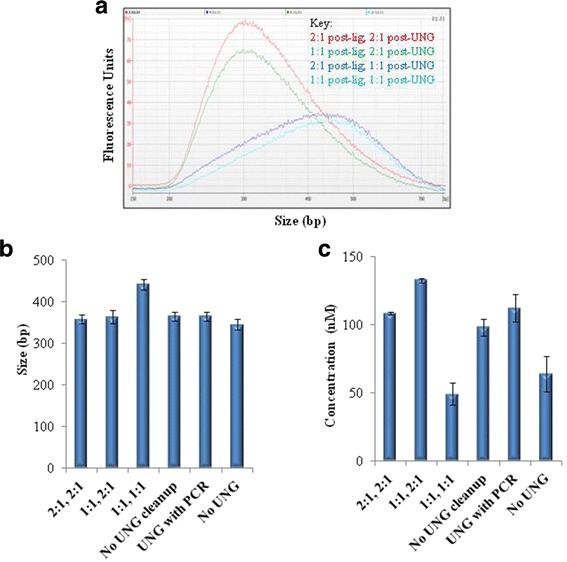
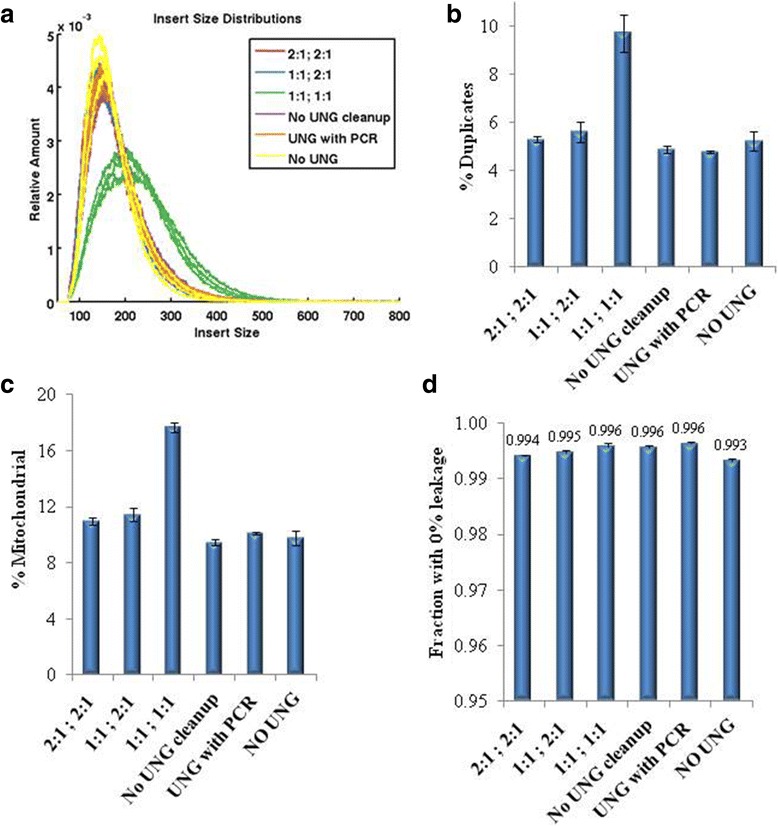
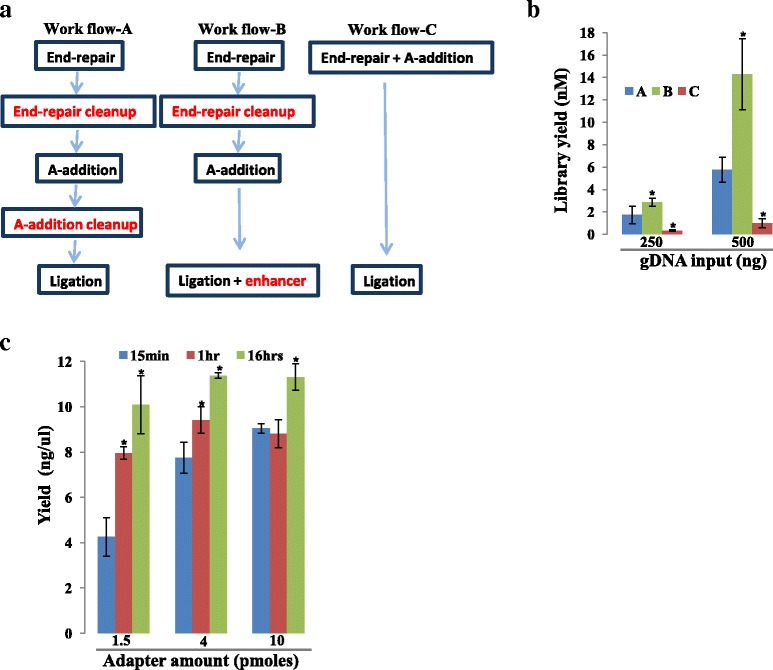
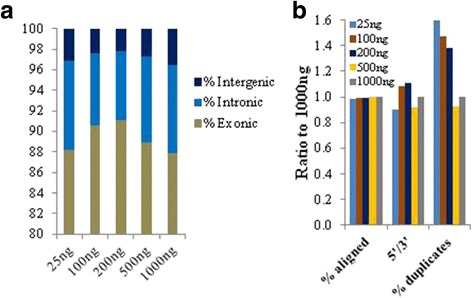
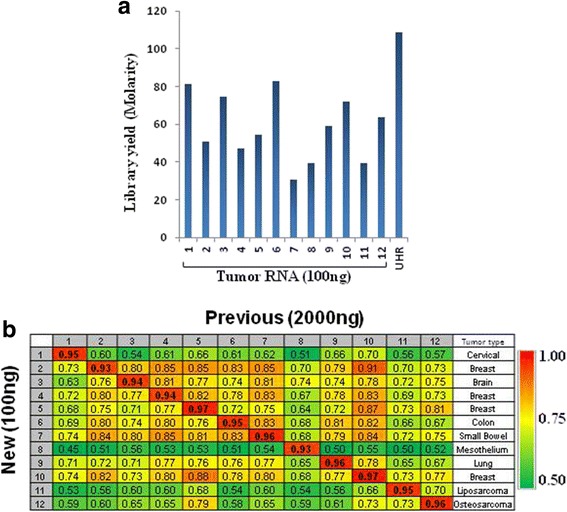
Background: RNA-Sequencing (RNA-seq) is now commonly used to reveal quantitative spatiotemporal snapshots of the transcriptome, the structures of transcripts (splice variants and fusions) and landscapes of expressed mutations. However, standard approaches for library construction typically require relatively high amounts of input RNA, are labor intensive, and are time consuming. METHODS: Here, we report the outcome of a systematic effort to optimize and streamline steps in strand-specific RNA-seq library construction. RESULTS: This work has resulted in the identification of an optimized messenger RNA isolation protocol, a potent reverse transcriptase for cDNA synthesis, and an efficient chemistry and a simplified formulation of library construction reagents. We also present an optimization of bead-based purification and size selection designed to maximize the recovery of cDNA fragments.
Conclusions: These developments have allowed us to assemble a rapid high throughput pipeline that produces high quality data from amounts of total RNA as low as 25 ng. While the focus of this study is on RNA-seq sample preparation, some of these developments are also relevant to other next-generation sequencing library types.
Keywords: Ampure XP magnetic beads; Illumina; Library construction; Ligation; Next-generation sequencing; RNA-seq; Reverse transcriptase; Strand-specific; Uracil DNA N-Glycosylase; dUTP; mRNA.
Conflict of interest statement
Ethics approval and consent to participate
Some of the patient samples used in this study are part of the BC Cancer Agency’s Personalized Oncogenomics project [14], which was approved by the University of British Columbia Research Ethics Committee (REB# H12–00137). Sample use was according to the written consent by each patient. Patient identity was made anonymous and sequence data and analyses thereafter were maintained within a secure computing environment.
Human promyelocytic Leukemia cell line (HL60) was purchased from Cedarlane Laboratories LTD.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
-
- Engler MJ, Richardson CC. DNA ligases. The Enzymes (Boyer PD, ed.), Academic Press, Inc, New York. 1982;3–29.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
