

Identification of pathogenic gene mutations in LMNA and MYBPC3 that alter RNA splicing
- PMID: 28679633
- PMCID: PMC5528995
- DOI: 10.1073/pnas.1707741114
Identification of pathogenic gene mutations in LMNA and MYBPC3 that alter RNA splicing
Abstract
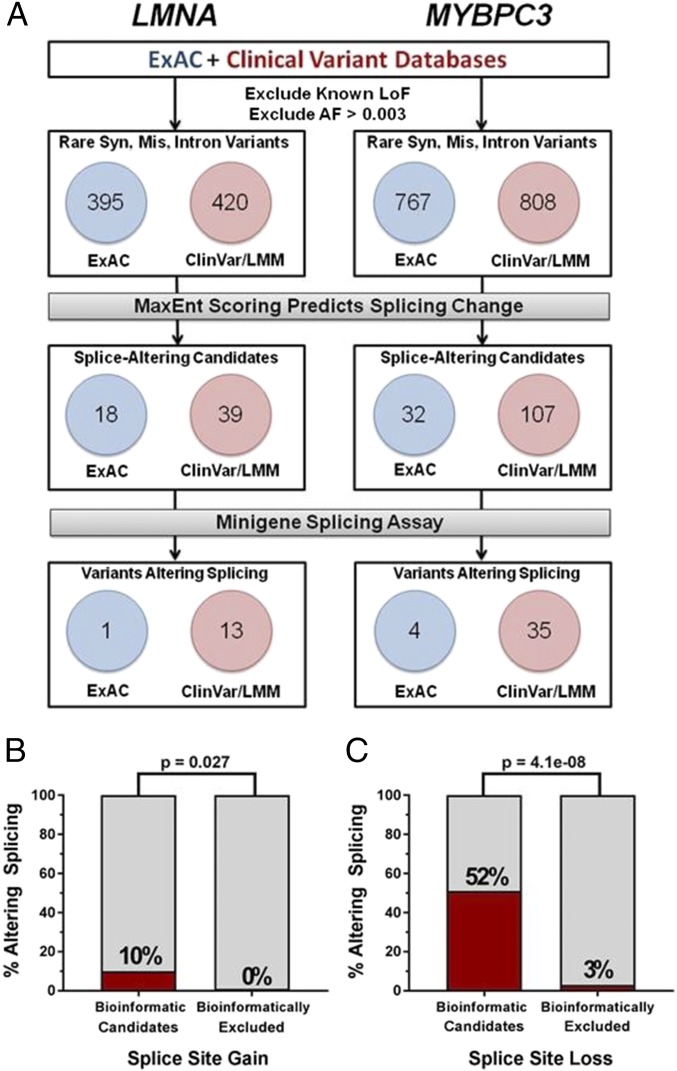
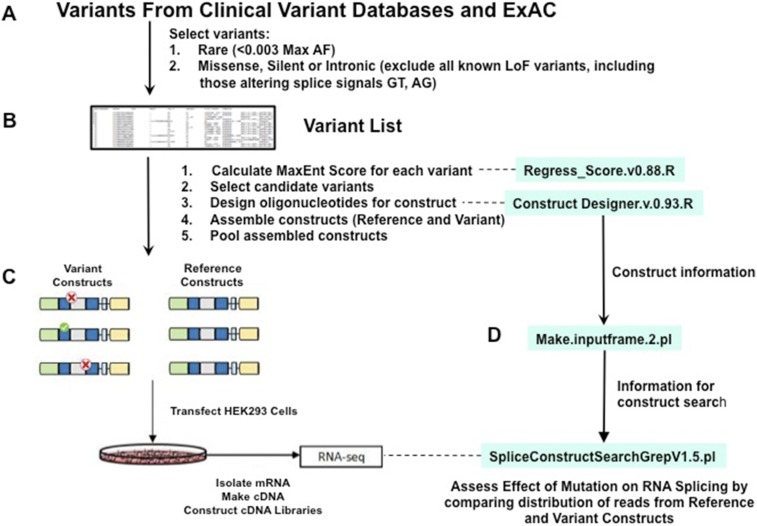
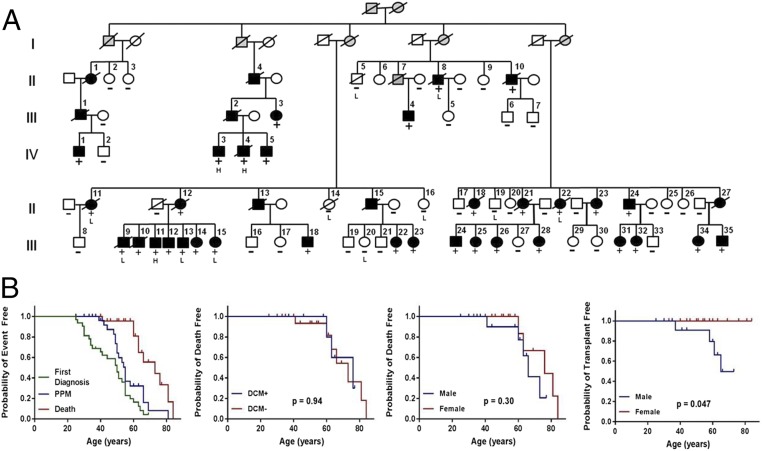
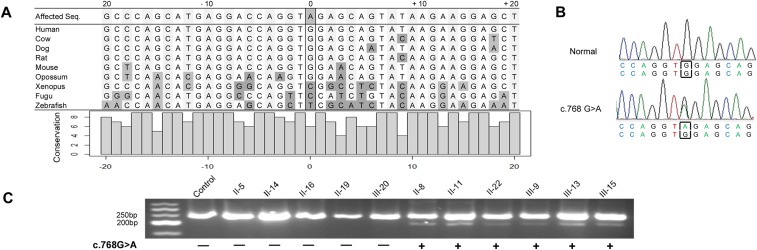
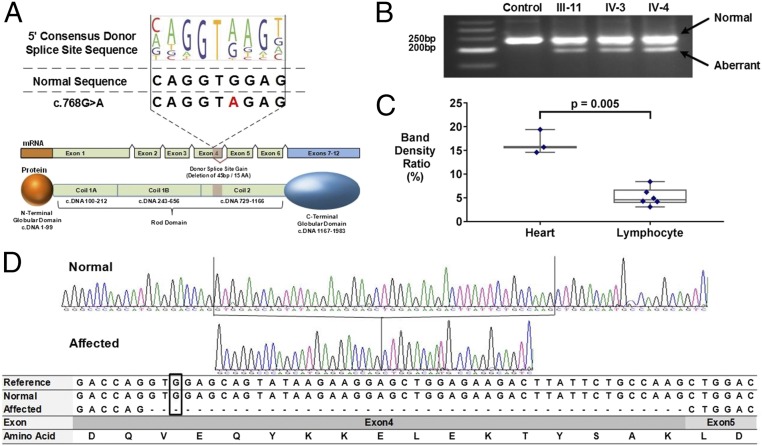
Genetic variants that cause haploinsufficiency account for many autosomal dominant (AD) disorders. Gene-based diagnosis classifies variants that alter canonical splice signals as pathogenic, but due to imperfect understanding of RNA splice signals other variants that may create or eliminate splice sites are often clinically classified as variants of unknown significance (VUS). To improve recognition of pathogenic splice-altering variants in AD disorders, we used computational tools to prioritize VUS and developed a cell-based minigene splicing assay to confirm aberrant splicing. Using this two-step procedure we evaluated all rare variants in two AD cardiomyopathy genes, lamin A/C (LMNA) and myosin binding protein C (MYBPC3). We demonstrate that 13 LMNA and 35 MYBPC3 variants identified in cardiomyopathy patients alter RNA splicing, representing a 50% increase in the numbers of established damaging splice variants in these genes. Over half of these variants are annotated as VUS by clinical diagnostic laboratories. Familial analyses of one variant, a synonymous LMNA VUS, demonstrated segregation with cardiomyopathy affection status and altered cardiac LMNA splicing. Application of this strategy should improve diagnostic accuracy and variant classification in other haploinsufficient AD disorders.
Keywords: LMNA; MYBPC3; VUS; cardiomyopathy; splicing.
Conflict of interest statement
Conflict of interest statement: C.E.S. and J.G.S. are founders of and own shares in Myokardia Inc., a startup company that is developing therapeutics that target the sarcomere.
Figures

References
-
- Medicine M-NIoG 2017. Online Mendelian Inheritance in Man (Johns Hopkins Univ, Baltimore)
-
- Richards S, et al. ACMG Laboratory Quality Assurance Committee Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. - PMC - PubMed
-
- Krawczak M, et al. Single base-pair substitutions in exon-intron junctions of human genes: Nature, distribution, and consequences for mRNA splicing. Hum Mutat. 2007;28:150–158. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
