

A salt-sensing kinase in T lymphocytes, SGK1, drives hypertension and hypertensive end-organ damage
- PMID: 28679951
- PMCID: PMC5499362
- DOI: 10.1172/jci.insight.92801
A salt-sensing kinase in T lymphocytes, SGK1, drives hypertension and hypertensive end-organ damage
Abstract
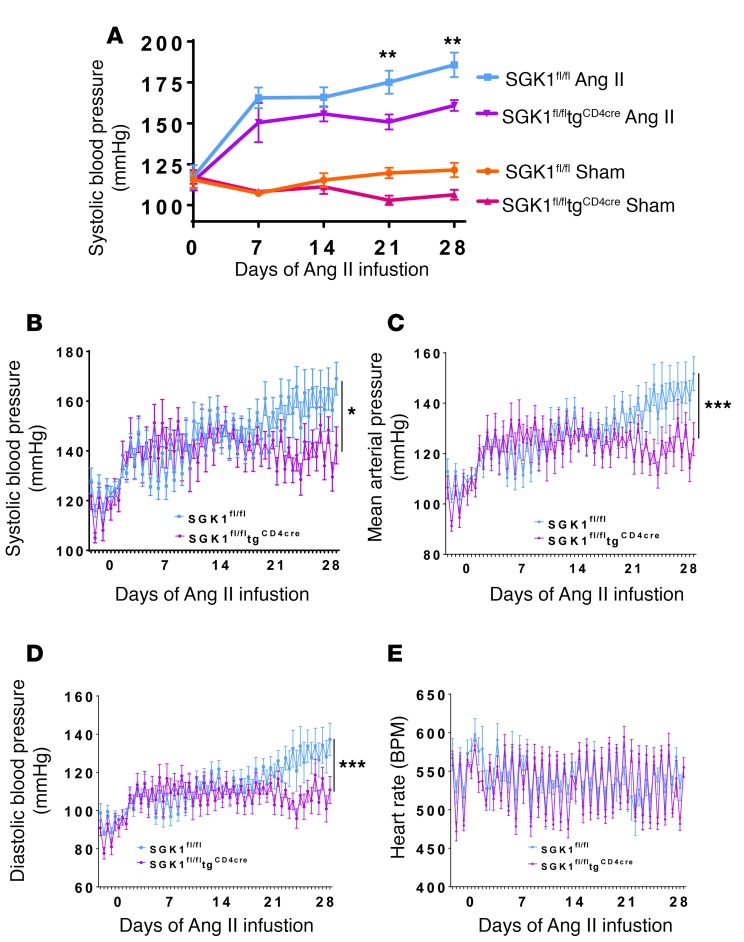
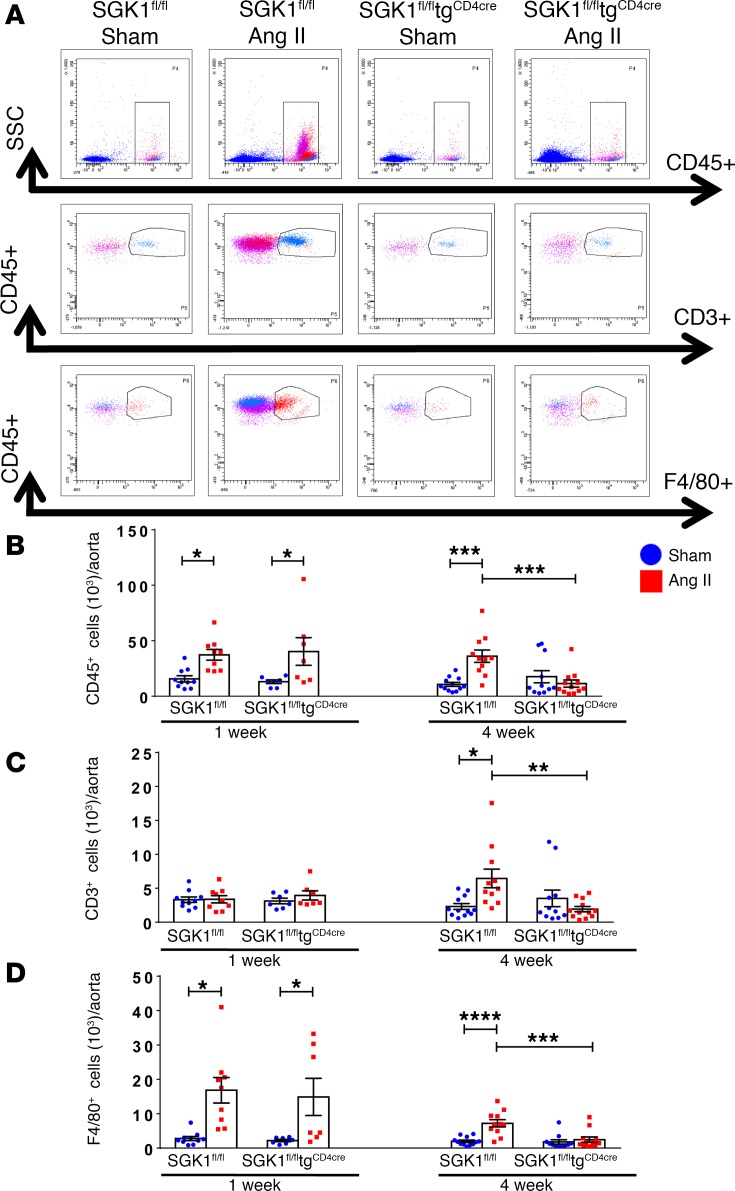
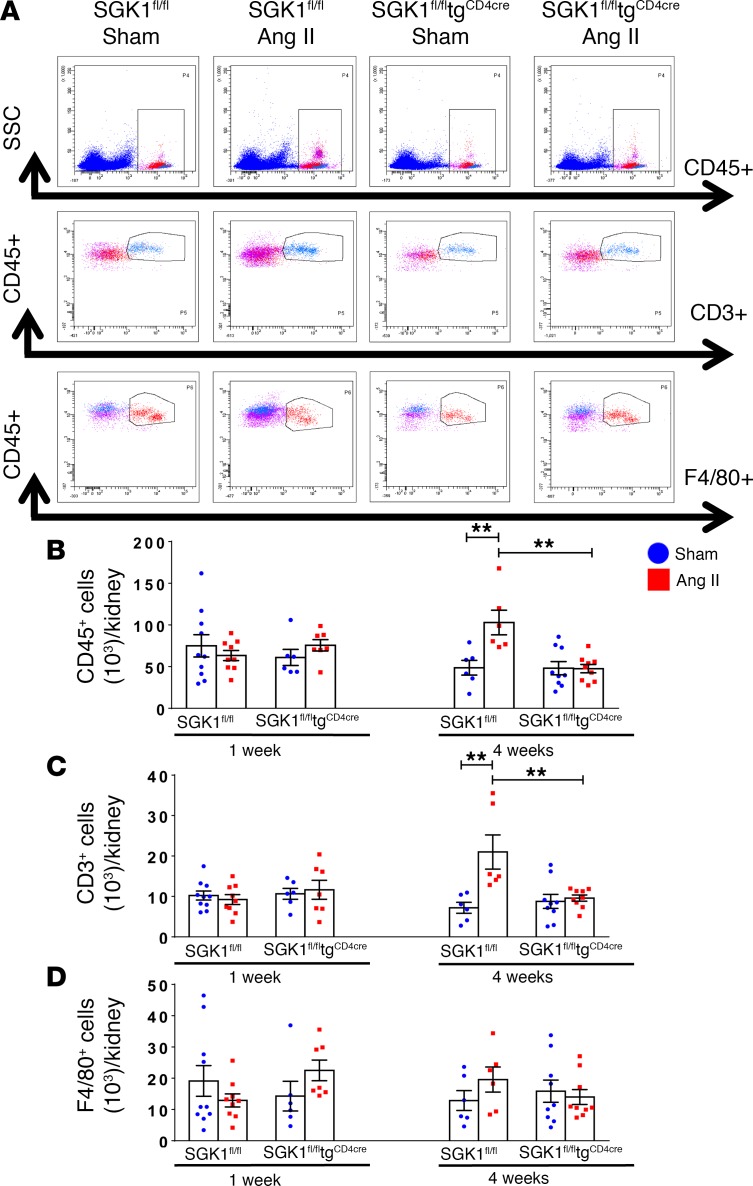
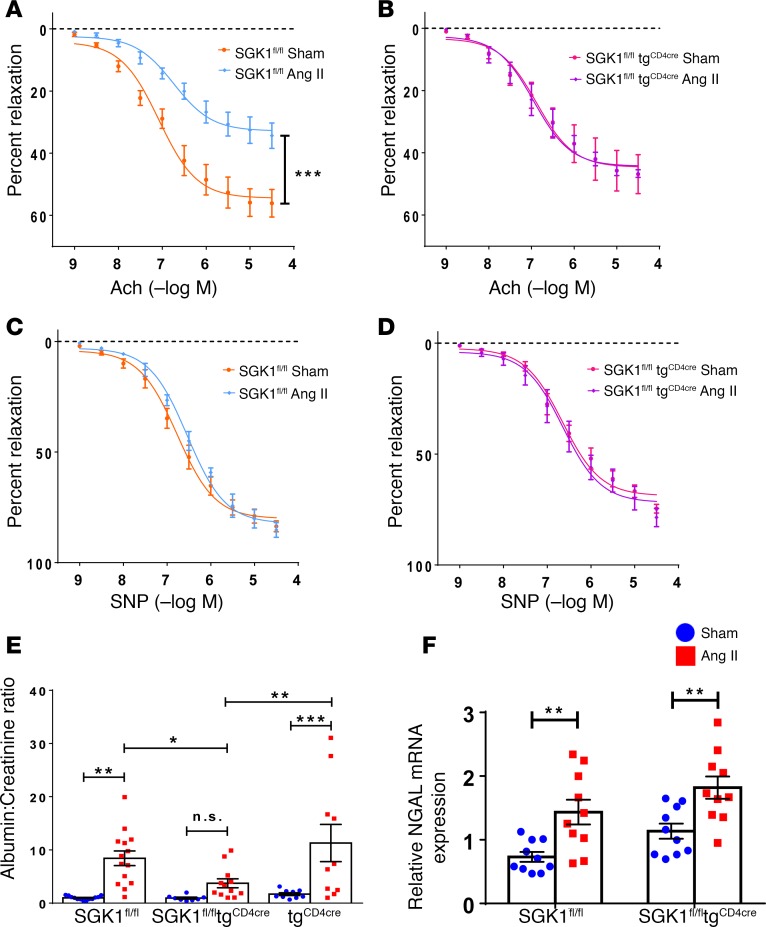
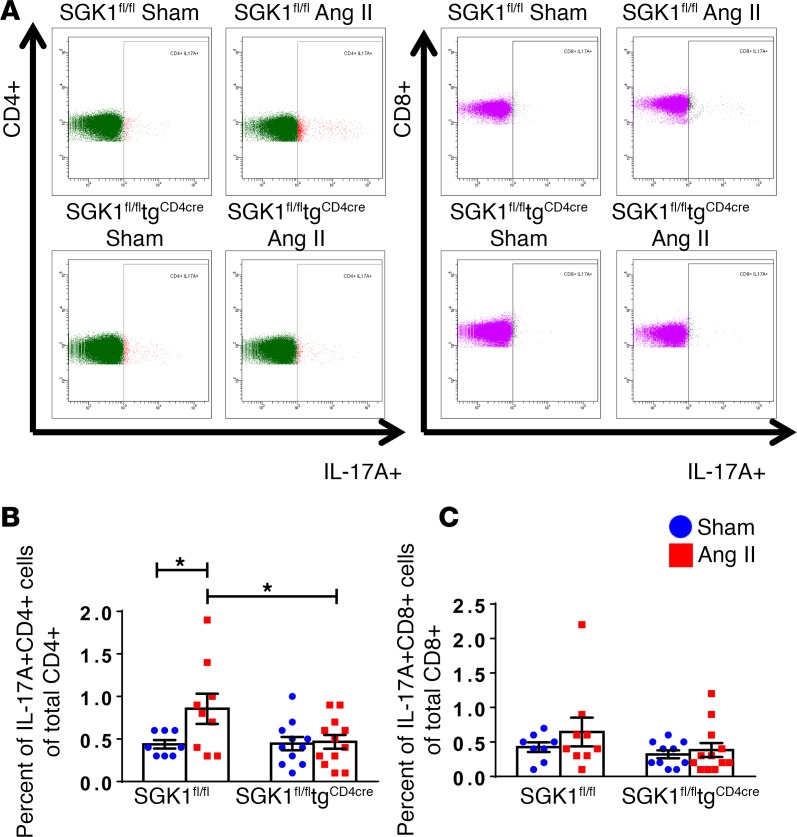
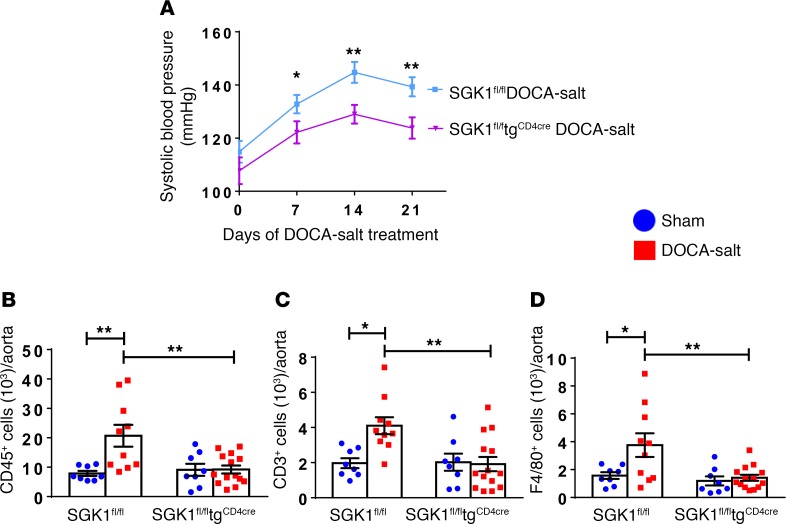
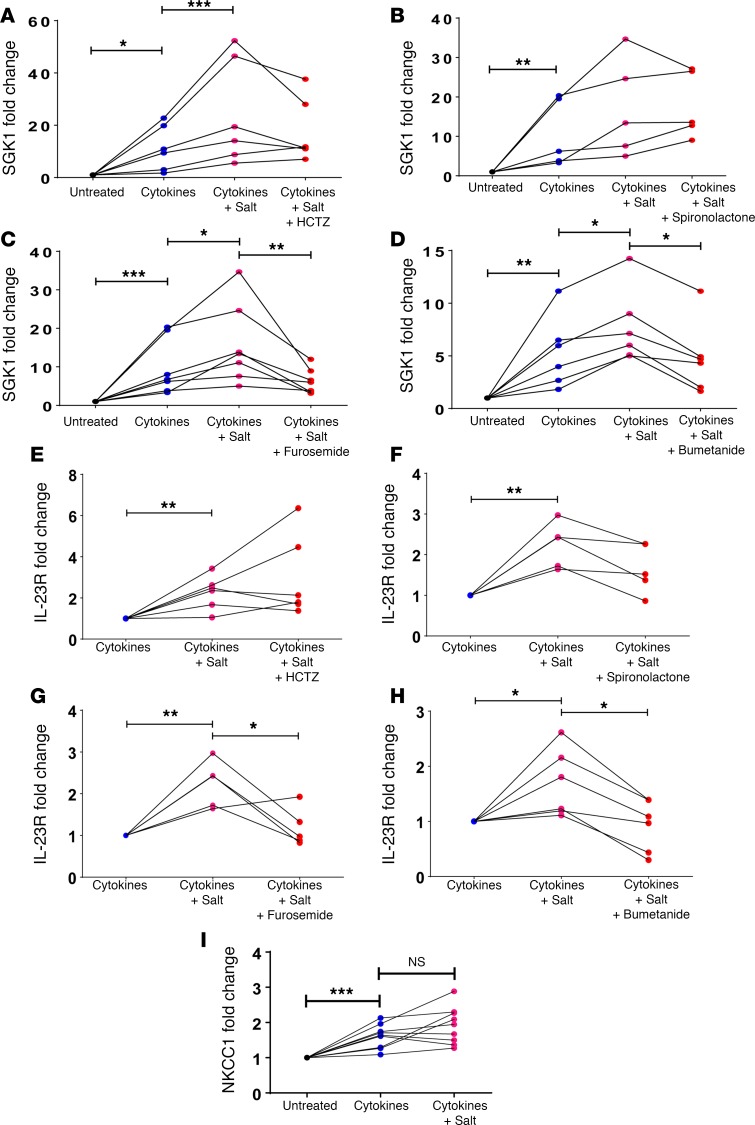
We previously showed that angiotensin II (Ang II) increases T cell production of IL-17A, and that mice deficient in IL-17A have blunted hypertension and attenuated renal and vascular dysfunction. It was recently shown that salt enhances IL-17A production from CD4+ T cells via a serum- and glucocorticoid-regulated kinase 1-dependent (SGK1-dependent) pathway. Thus, we tested the hypothesis that SGK1 signaling in T cells promotes hypertension and contributes to end-organ damage. We show that loss of T cell SGK1 results in a blunted hypertensive response to Ang II infusion by 25 mmHg. Importantly, renal and vascular inflammation is abrogated in these mice compared with control mice. Furthermore, mice lacking T cell SGK1 are protected from Ang II-induced endothelial dysfunction and renal injury. Loss of T cell SGK1 also blunts blood pressure and vascular inflammation in response to deoxycorticosterone acetate-salt (DOCA-salt) hypertension. Finally, we demonstrate that the Na+-K+-2Cl- cotransporter 1 (NKCC1) is upregulated in Th17 cells and is necessary for the salt-induced increase in SGK1 and the IL-23 receptor. These studies demonstrate that T cell SGK1 and NKCC1 may be novel therapeutic targets for the treatment of hypertension and identify a potentially new mechanism by which salt contributes to hypertension.
Keywords: Adaptive immunity; Cardiology; Cardiovascular disease; Hypertension; Inflammation.
Conflict of interest statement
Figures
Similar articles
-
Inflammation in Salt-Sensitive Hypertension and Renal Damage.Curr Hypertens Rep. 2018 Oct 30;20(12):103. doi: 10.1007/s11906-018-0903-x. Curr Hypertens Rep. 2018. PMID: 30377822 Free PMC article. Review.
-
Adipose c-Jun NH2-terminal kinase promotes angiotensin II-induced and deoxycorticosterone acetate salt-induced hypertension and vascular dysfunction by inhibition of adiponectin production and activation of SGK1 in mice.J Hypertens. 2024 May 1;42(5):856-872. doi: 10.1097/HJH.0000000000003649. Epub 2023 Dec 20. J Hypertens. 2024. PMID: 38164960
-
Inhibition of Interleukin 17-A but not Interleukin-17F Signaling Lowers Blood Pressure and Reduces End-organ Inflammation in Angiotensin II-induced Hypertension.JACC Basic Transl Sci. 2016 Dec;1(7):606-616. doi: 10.1016/j.jacbts.2016.07.009. Epub 2016 Nov 16. JACC Basic Transl Sci. 2016. PMID: 28280792 Free PMC article.
-
SGK1-FoxO1 Signaling Pathway Mediates Th17/Treg Imbalance and Target Organ Inflammation in Angiotensin II-Induced Hypertension.Front Physiol. 2018 Nov 15;9:1581. doi: 10.3389/fphys.2018.01581. eCollection 2018. Front Physiol. 2018. PMID: 30524295 Free PMC article.
-
Interleukin 17A: Key Player in the Pathogenesis of Hypertension and a Potential Therapeutic Target.Curr Hypertens Rep. 2021 Mar 5;23(3):13. doi: 10.1007/s11906-021-01128-7. Curr Hypertens Rep. 2021. PMID: 33666761 Free PMC article. Review.
Cited by
-
ENaC in Salt-Sensitive Hypertension: Kidney and Beyond.Curr Hypertens Rep. 2020 Aug 27;22(9):69. doi: 10.1007/s11906-020-01067-9. Curr Hypertens Rep. 2020. PMID: 32852643 Free PMC article. Review.
-
Responses Triggered by the Immune System in Hypertensive Conditions and Repercussions on Target Organ Damage: A Review.Curr Cardiol Rev. 2023;19(2):e200922208959. doi: 10.2174/1573403X18666220920090632. Curr Cardiol Rev. 2023. PMID: 36125837 Free PMC article. Review.
-
Inflammation in Salt-Sensitive Hypertension and Renal Damage.Curr Hypertens Rep. 2018 Oct 30;20(12):103. doi: 10.1007/s11906-018-0903-x. Curr Hypertens Rep. 2018. PMID: 30377822 Free PMC article. Review.
-
Immune and inflammatory mechanisms in hypertension.Nat Rev Cardiol. 2024 Jun;21(6):396-416. doi: 10.1038/s41569-023-00964-1. Epub 2024 Jan 3. Nat Rev Cardiol. 2024. PMID: 38172242 Review.
-
Hypertension and human immunodeficiency virus: A paradigm for epithelial sodium channels?Front Cardiovasc Med. 2022 Aug 25;9:968184. doi: 10.3389/fcvm.2022.968184. eCollection 2022. Front Cardiovasc Med. 2022. PMID: 36093171 Free PMC article. Review.
References
-
- Nwankwo T, Yoon SS, Burt V, Gu Q. 2013. Hypertension Among Adults in the United States: National Health and Nutrition Examination Survey, 2011-2012. NCHS Data Brief. 2013;(133):1–8. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
