

SOCS1 is a negative regulator of metabolic reprogramming during sepsis
- PMID: 28679957
- PMCID: PMC5499360
- DOI: 10.1172/jci.insight.92530
SOCS1 is a negative regulator of metabolic reprogramming during sepsis
Abstract
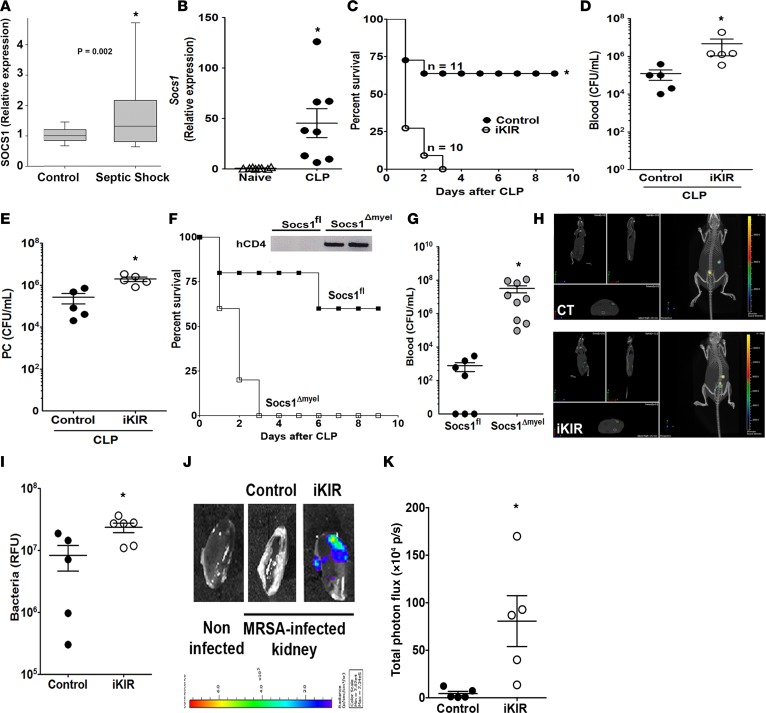
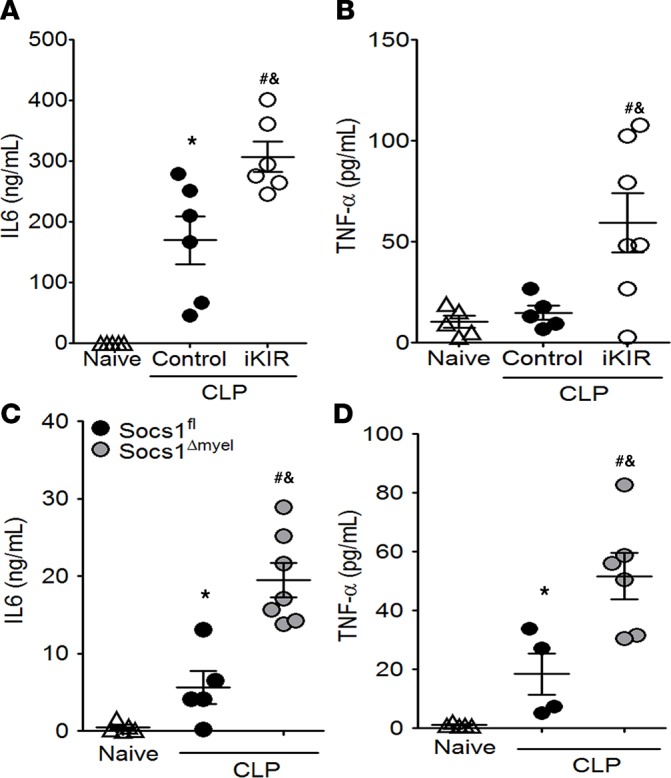
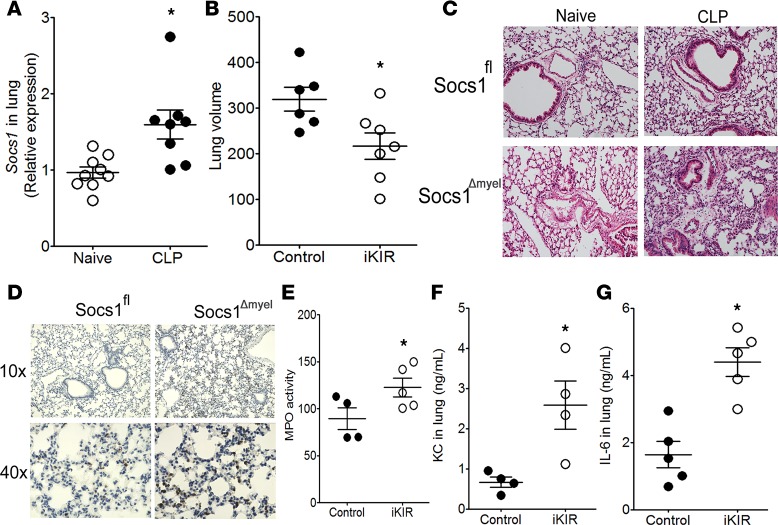
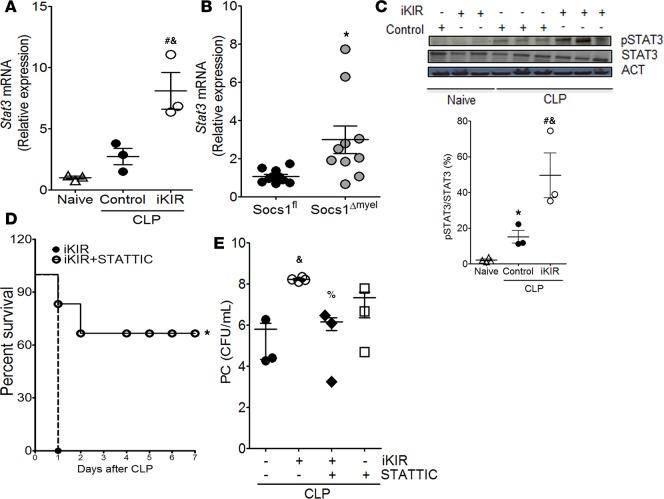
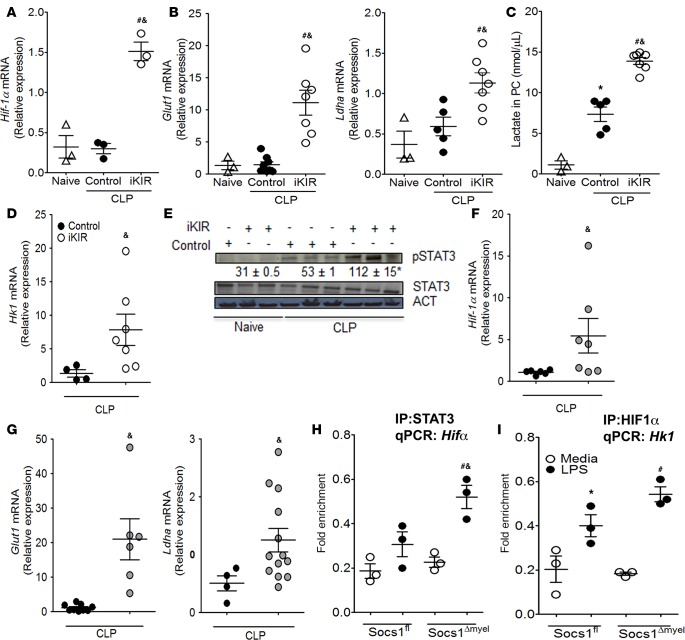
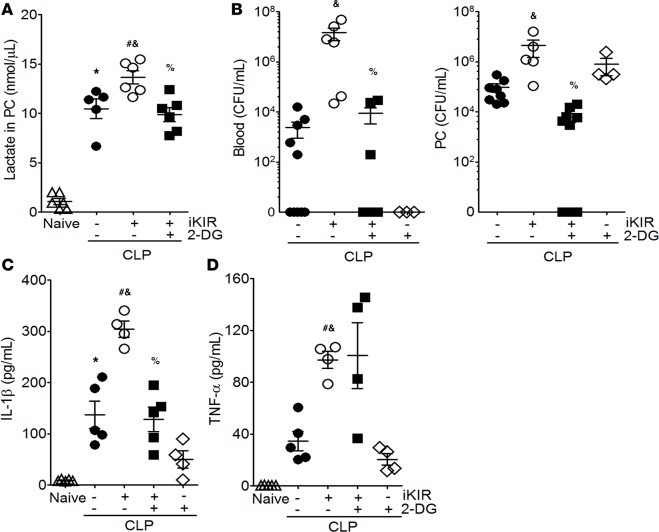
Sepsis can induce an overwhelming systemic inflammatory response, resulting in organ damage and death. Suppressor of cytokine signaling 1 (SOCS1) negatively regulates signaling by cytokine receptors and Toll-like receptors (TLRs). However, the cellular targets and molecular mechanisms for SOCS1 activity during polymicrobial sepsis are unknown. To address this, we utilized a cecal ligation and puncture (CLP) model for sepsis; C57BL/6 mice subjected to CLP were then treated with a peptide (iKIR) that binds the SOCS1 kinase inhibitory region (KIR) and blocks its activity. Treatment with iKIR increased CLP-induced mortality, bacterial burden, and inflammatory cytokine production. Myeloid cell-specific SOCS1 deletion (Socs1Δmyel) mice were also more susceptible to sepsis, demonstrating increased mortality, higher bacterial loads, and elevated inflammatory cytokines, compared with Socs1fl littermate controls. These effects were accompanied by macrophage metabolic reprograming, as evidenced by increased lactic acid production and elevated expression of the glycolytic enzymes hexokinase, lactate dehydrogenase A, and glucose transporter 1 in septic Socs1Δmyel mice. Upregulation was dependent on the STAT3/HIF-1α/glycolysis axis, and blocking glycolysis ameliorated increased susceptibility to sepsis in iKIR-treated CLP mice. These results reveal a role of SOCS1 as a regulator of metabolic reprograming that prevents overwhelming inflammatory response and organ damage during sepsis.
Keywords: Immunology; Metabolism.
Conflict of interest statement
Figures
Similar articles
-
Enhanced Glycolytic Metabolism Contributes to Cardiac Dysfunction in Polymicrobial Sepsis.J Infect Dis. 2017 May 1;215(9):1396-1406. doi: 10.1093/infdis/jix138. J Infect Dis. 2017. PMID: 28368517 Free PMC article.
-
[Study on the expression of suppressor of cytokine signaling-1 in liver and spleen of septic mice].Zhongguo Wei Zhong Bing Ji Jiu Yi Xue. 2007 Oct;19(10):606-9. Zhongguo Wei Zhong Bing Ji Jiu Yi Xue. 2007. PMID: 17945083 Chinese.
-
Poly(I:C) Priming Exacerbates Cecal Ligation and Puncture-Induced Polymicrobial Sepsis in Mice.Inflammation. 2018 Feb;41(1):328-336. doi: 10.1007/s10753-017-0690-6. Inflammation. 2018. PMID: 29127663 Free PMC article.
-
Delayed Mitogen-Activated Protein Kinase/Extracellular Signal-Regulated Kinase Inhibition by Trametinib Attenuates Systemic Inflammatory Responses and Multiple Organ Injury in Murine Sepsis.Crit Care Med. 2016 Aug;44(8):e711-20. doi: 10.1097/CCM.0000000000001672. Crit Care Med. 2016. PMID: 27031380 Free PMC article.
-
Lactate Is a Metabolic Mediator That Shapes Immune Cell Fate and Function.Front Physiol. 2021 Oct 18;12:688485. doi: 10.3389/fphys.2021.688485. eCollection 2021. Front Physiol. 2021. PMID: 34733170 Free PMC article. Review.
Cited by
-
Acyloxyacyl hydrolase promotes pulmonary defense by preventing alveolar macrophage tolerance.PLoS Pathog. 2023 Jul 27;19(7):e1011556. doi: 10.1371/journal.ppat.1011556. eCollection 2023 Jul. PLoS Pathog. 2023. PMID: 37498977 Free PMC article.
-
Exosomal mediators in sepsis and inflammatory organ injury: unraveling the role of exosomes in intercellular crosstalk and organ dysfunction.Mil Med Res. 2024 Apr 22;11(1):24. doi: 10.1186/s40779-024-00527-6. Mil Med Res. 2024. PMID: 38644472 Free PMC article. Review.
-
Cellular and Exosomal Regulations of Sepsis-Induced Metabolic Alterations.Int J Mol Sci. 2021 Aug 2;22(15):8295. doi: 10.3390/ijms22158295. Int J Mol Sci. 2021. PMID: 34361061 Free PMC article. Review.
-
SOCS-1 inhibition of type I interferon restrains Staphylococcus aureus skin host defense.PLoS Pathog. 2021 Mar 10;17(3):e1009387. doi: 10.1371/journal.ppat.1009387. eCollection 2021 Mar. PLoS Pathog. 2021. PMID: 33690673 Free PMC article.
-
IFI44 is an immune evasion biomarker for SARS-CoV-2 and Staphylococcus aureus infection in patients with RA.Front Immunol. 2022 Sep 15;13:1013322. doi: 10.3389/fimmu.2022.1013322. eCollection 2022. Front Immunol. 2022. PMID: 36189314 Free PMC article.
References
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
