

The Role of the Heat Shock Protein B8 (HSPB8) in Motoneuron Diseases
- PMID: 28680390
- PMCID: PMC5478700
- DOI: 10.3389/fnmol.2017.00176
The Role of the Heat Shock Protein B8 (HSPB8) in Motoneuron Diseases
Abstract
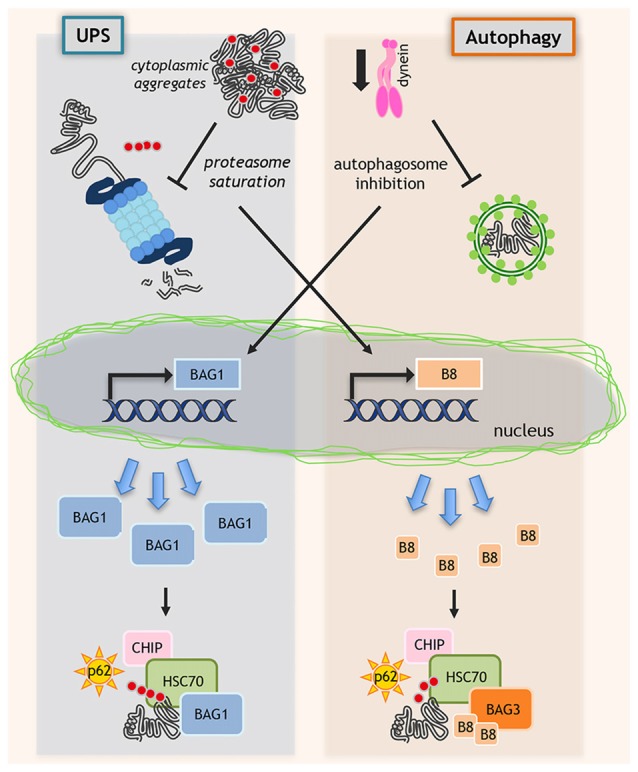
Amyotrophic lateral sclerosis (ALS) and spinal and bulbar muscular atrophy (SBMA) are two motoneuron diseases (MNDs) characterized by aberrant protein behavior in affected cells. In familial ALS (fALS) and in SBMA specific gene mutations lead to the production of neurotoxic proteins or peptides prone to misfold, which then accumulate in form of aggregates. Notably, some of these proteins accumulate into aggregates also in sporadic ALS (sALS) even if not mutated. To prevent proteotoxic stresses detrimental to cells, misfolded and/or aggregated proteins must be rapidly removed by the protein quality control (PQC) system. The small heat shock protein B8 (HSPB8) is a chaperone induced by harmful events, like proteasome inhibition. HSPB8 is expressed both in motoneuron and muscle cells, which are both targets of misfolded protein toxicity in MNDs. In ALS mice models, in presence of the mutant proteins, HSPB8 is upregulated both in spinal cord and muscle. HSPB8 interacts with the HSP70 co-chaperone BAG3 and enhances the degradation of misfolded proteins linked to sALS, or causative of fALS and of SBMA. HSPB8 acts by facilitating autophagy, thereby preventing misfolded protein accumulation in affected cells. BAG3 and BAG1 compete for HSP70-bound clients and target them for disposal to the autophagy or proteasome, respectively. Enhancing the selective targeting of misfolded proteins by HSPB8-BAG3-HSP70 to autophagy may also decrease their delivery to the proteasome by the BAG1-HSP70 complex, thereby limiting possible proteasome overwhelming. Thus, approaches aimed at potentiating HSPB8-BAG3 may contribute to the maintenance of proteostasis and may delay MNDs progression.
Keywords: HSPB8; amyotrophic lateral sclerosis; autophagy; chaperones; misfolded proteins; motoneuron diseases; proteasome; spinal and bulbar muscular atrophy.
Figures

References
-
- Al-Sarraj S., King A., Troakes C., Smith B., Maekawa S., Bodi I., et al. . (2011). p62 positive, TDP-43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72-linked FTLD and MND/ALS. Acta Neuropathol. 122, 691–702. 10.1007/s00401-011-0911-2 - DOI - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous