

Cell-cycle dynamics of chromosomal organization at single-cell resolution
- PMID: 28682332
- PMCID: PMC5567812
- DOI: 10.1038/nature23001
Cell-cycle dynamics of chromosomal organization at single-cell resolution
Abstract
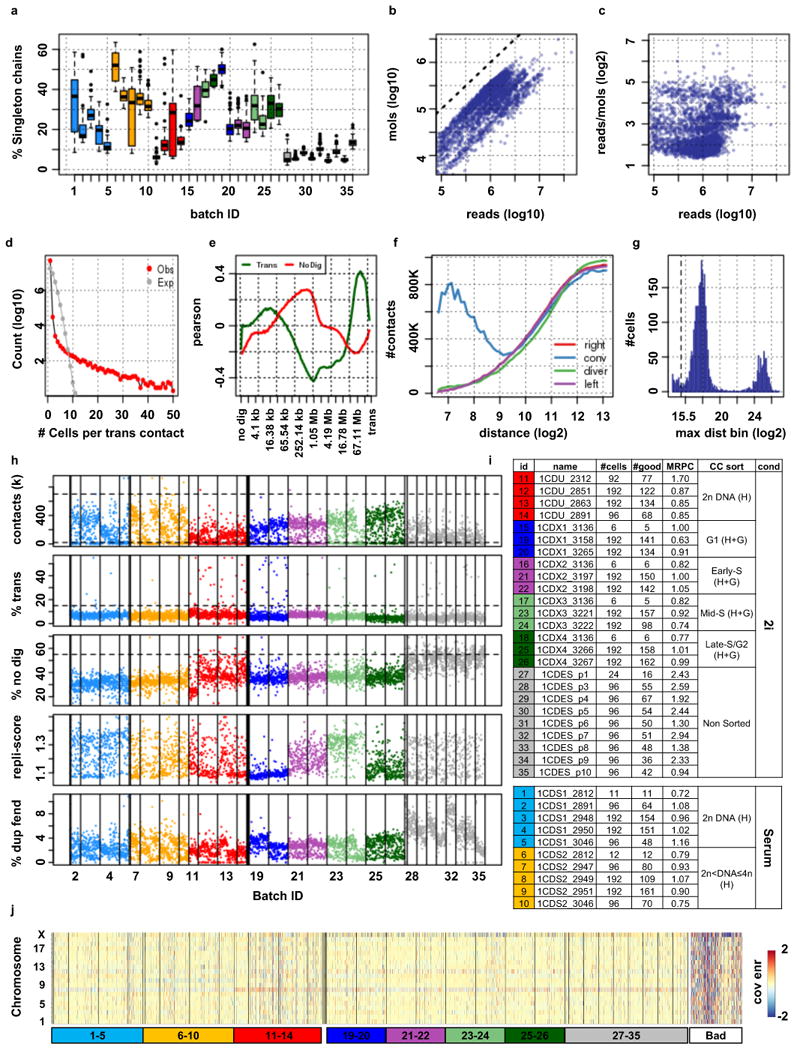
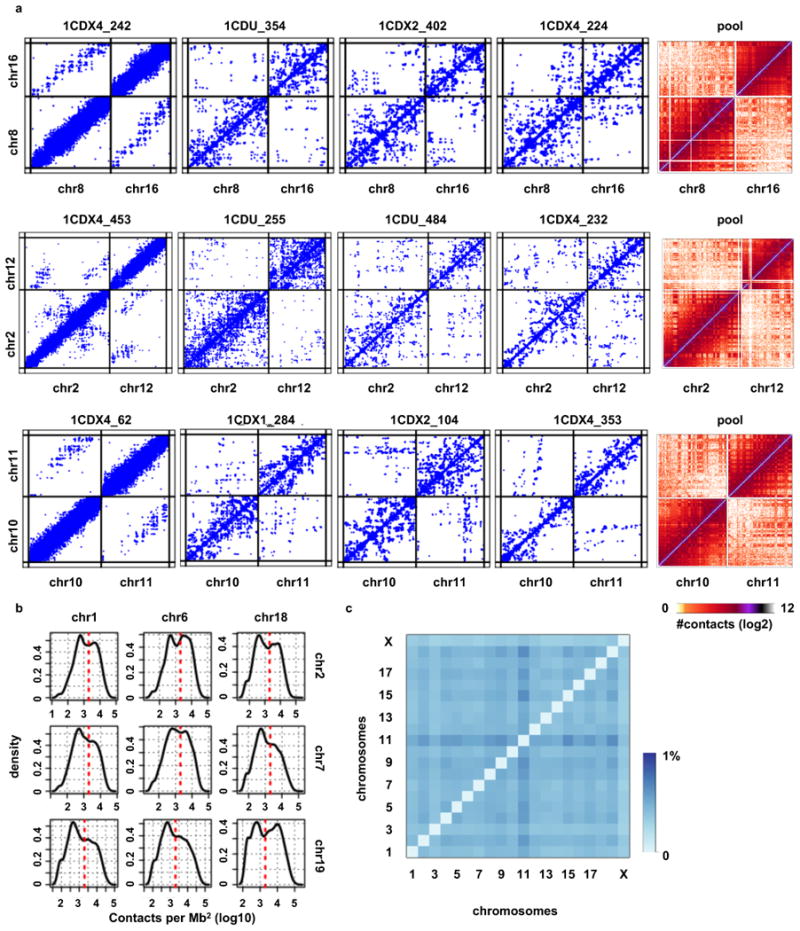
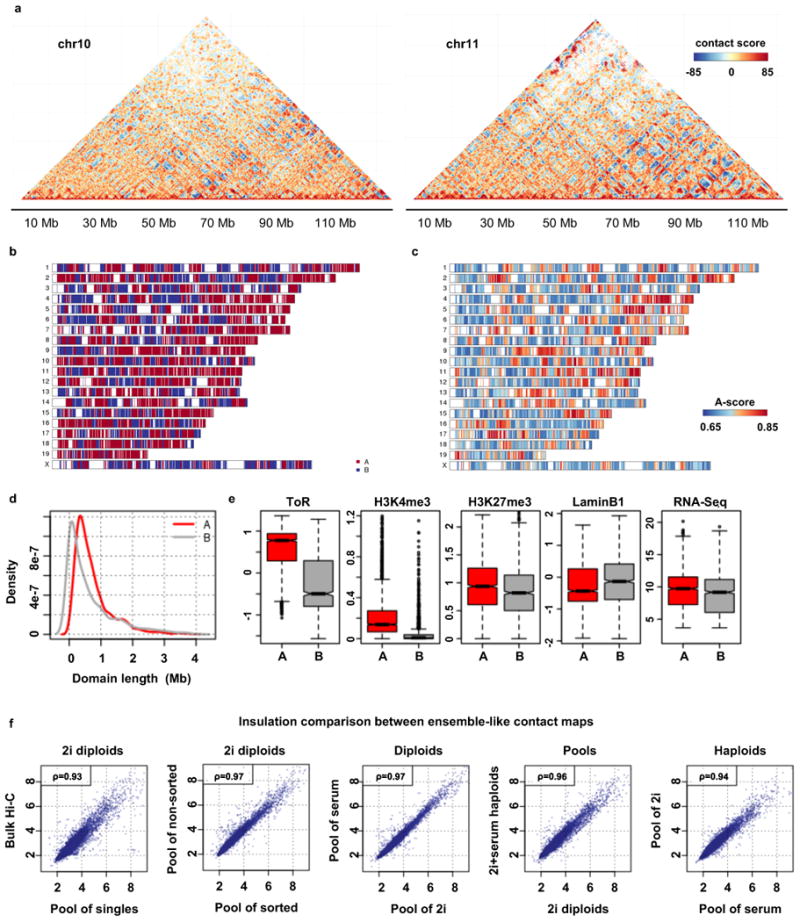
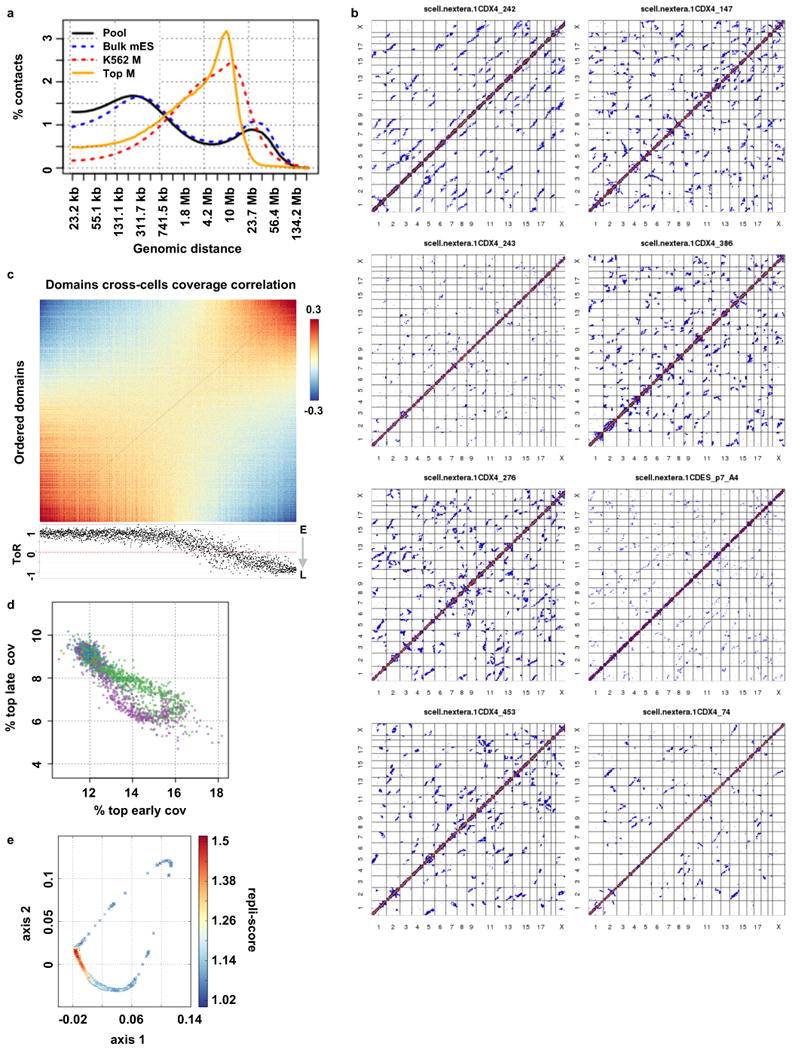
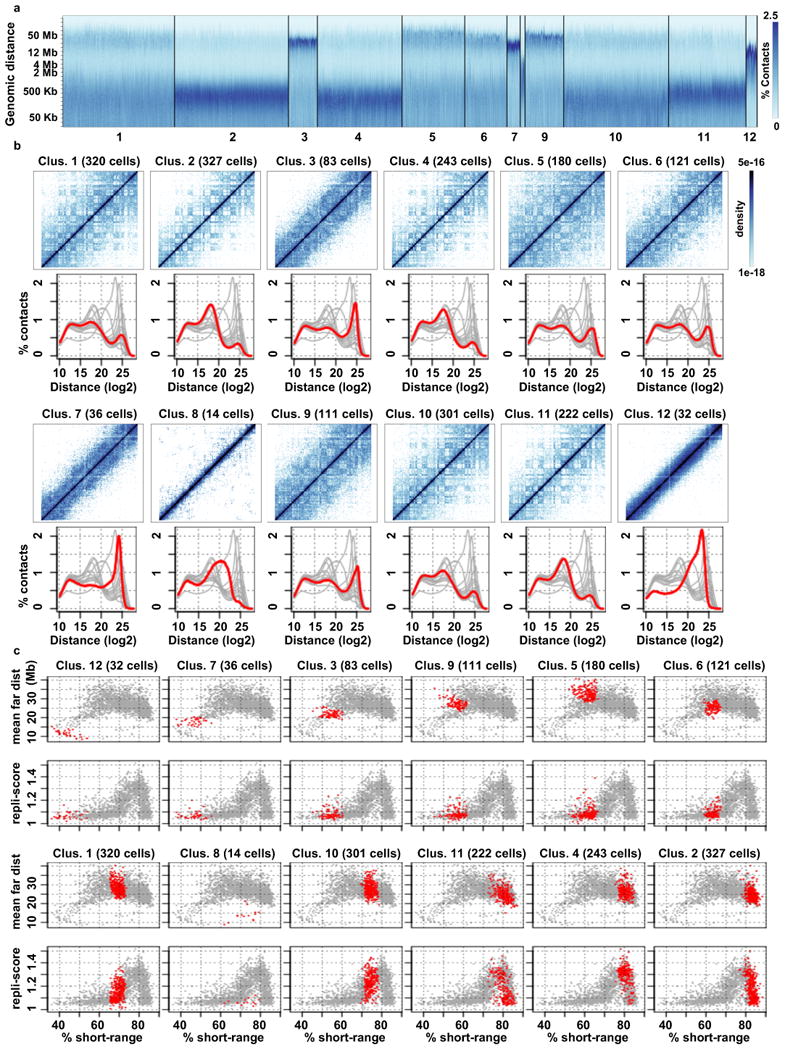
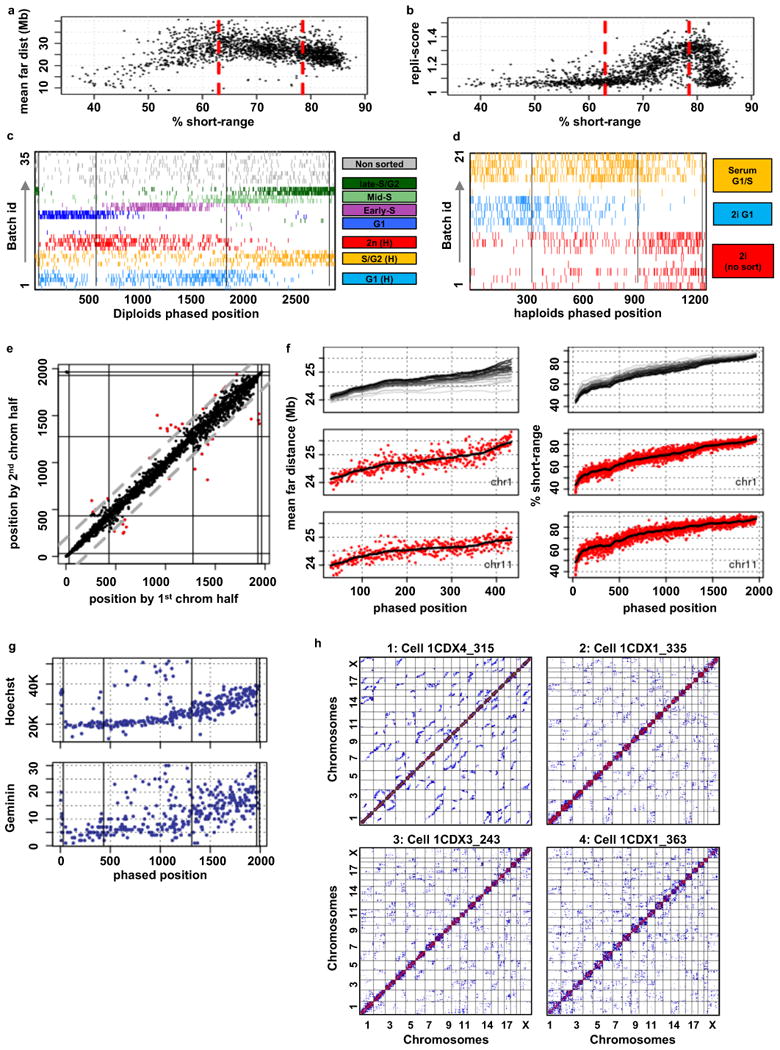
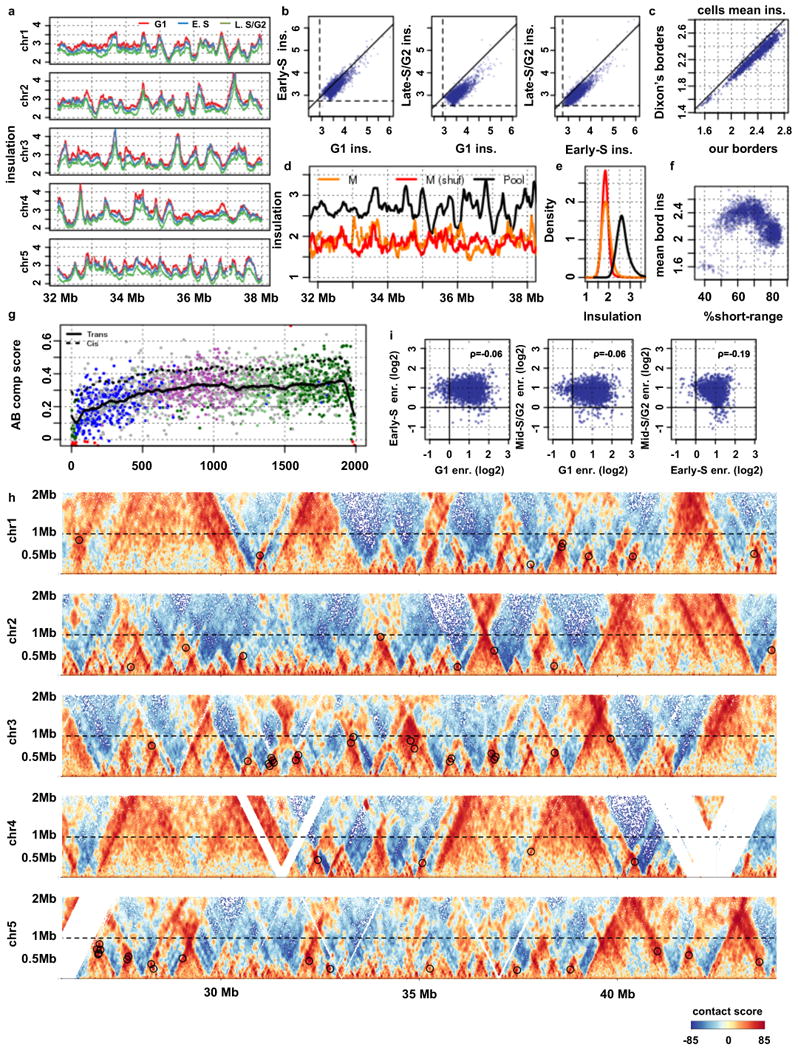
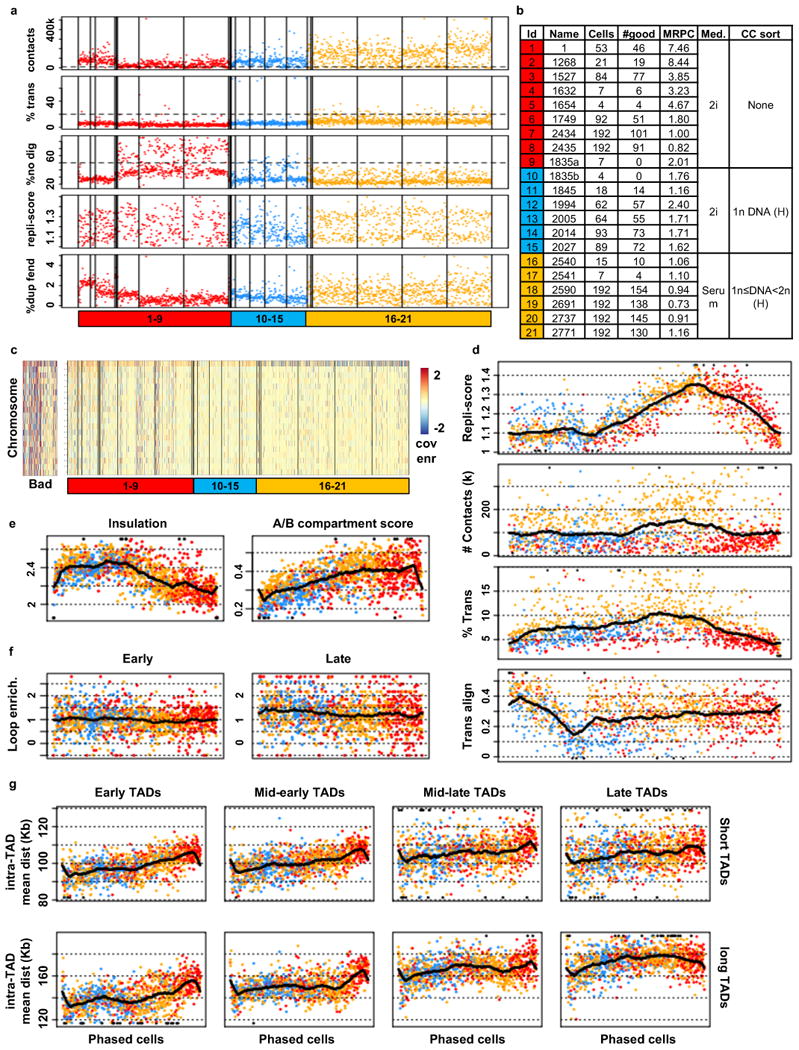
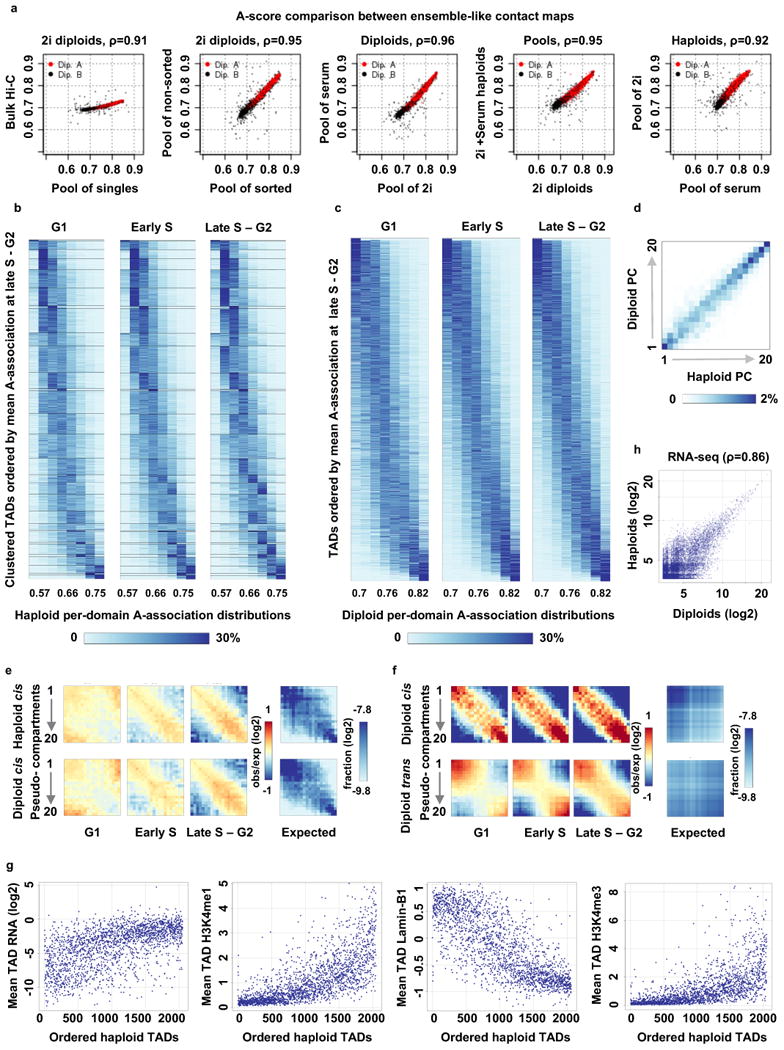
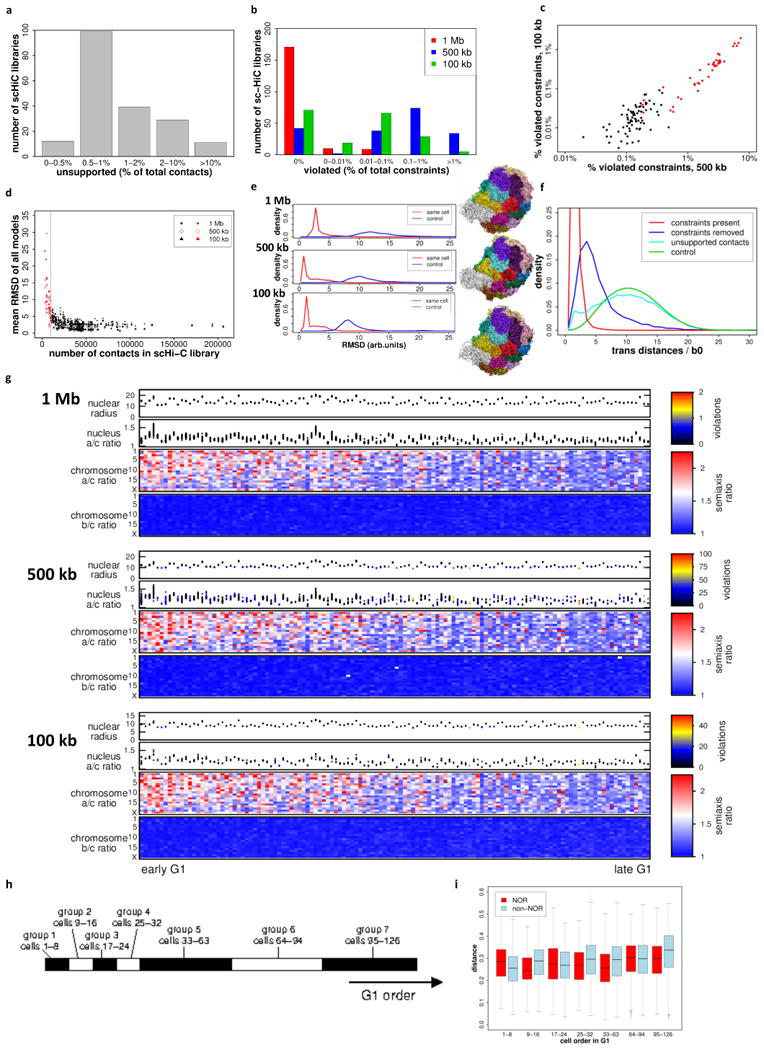
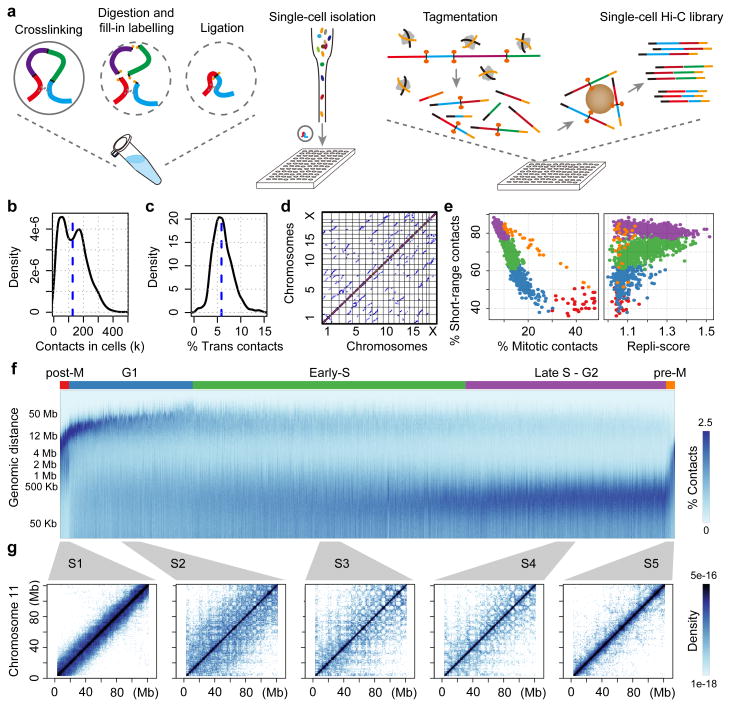
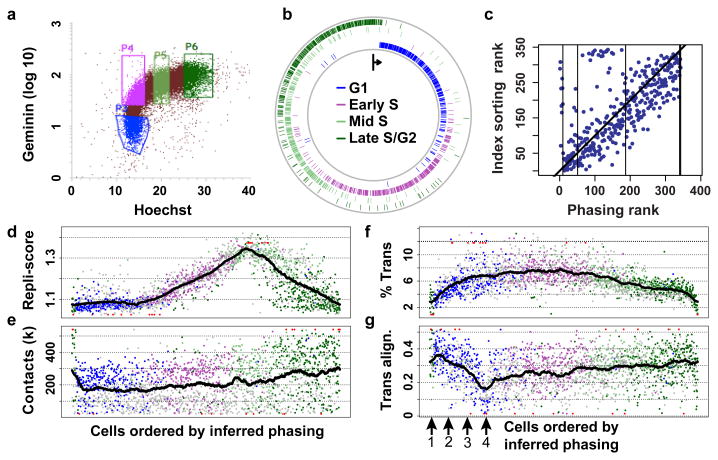
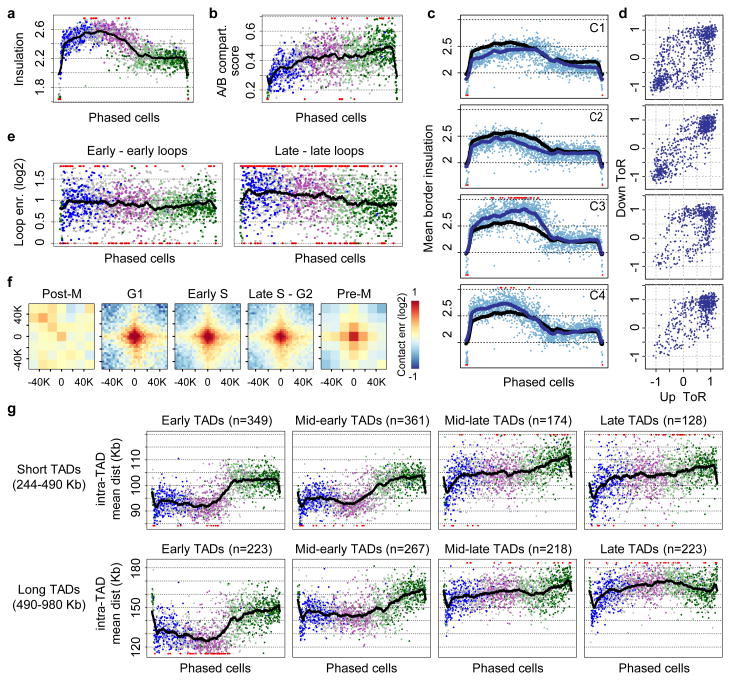
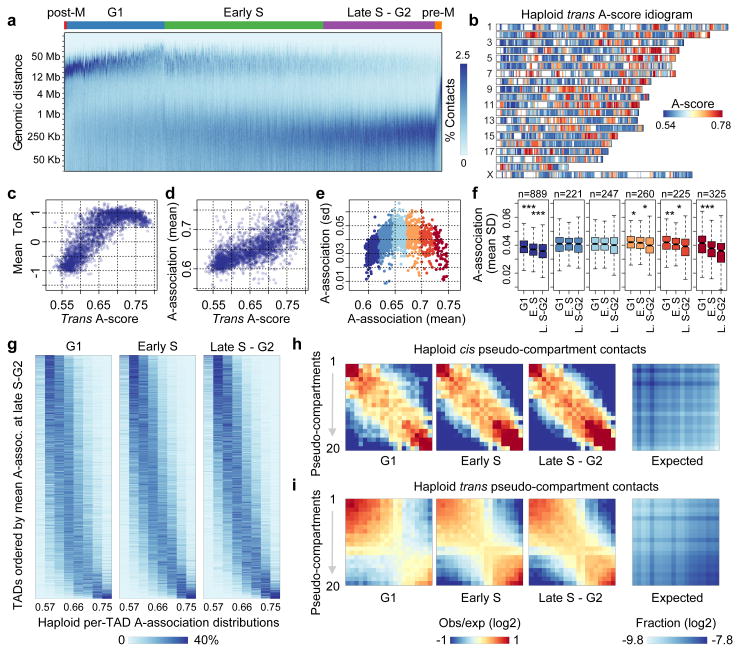
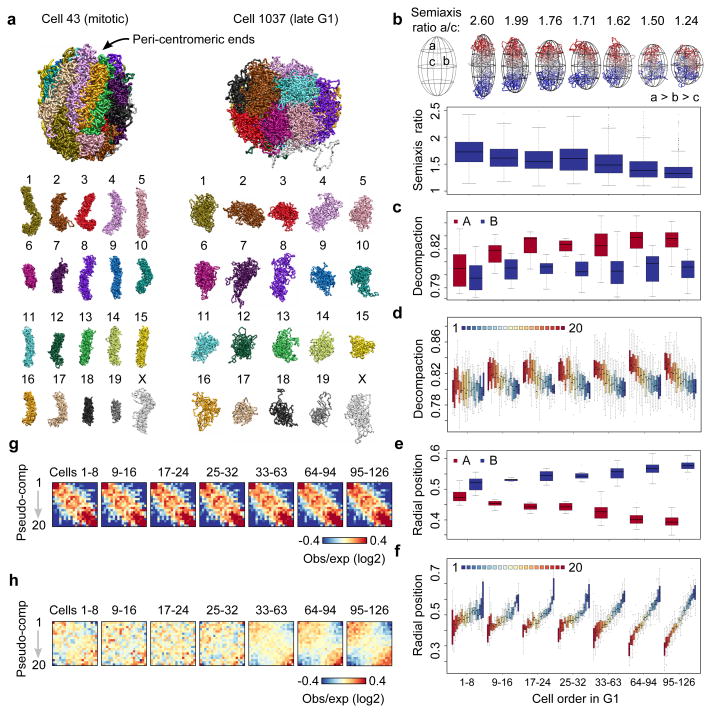
Chromosomes in proliferating metazoan cells undergo marked structural metamorphoses every cell cycle, alternating between highly condensed mitotic structures that facilitate chromosome segregation, and decondensed interphase structures that accommodate transcription, gene silencing and DNA replication. Here we use single-cell Hi-C (high-resolution chromosome conformation capture) analysis to study chromosome conformations in thousands of individual cells, and discover a continuum of cis-interaction profiles that finely position individual cells along the cell cycle. We show that chromosomal compartments, topological-associated domains (TADs), contact insulation and long-range loops, all defined by bulk Hi-C maps, are governed by distinct cell-cycle dynamics. In particular, DNA replication correlates with a build-up of compartments and a reduction in TAD insulation, while loops are generally stable from G1 to S and G2 phase. Whole-genome three-dimensional structural models reveal a radial architecture of chromosomal compartments with distinct epigenomic signatures. Our single-cell data therefore allow re-interpretation of chromosome conformation maps through the prism of the cell cycle.
Figures
Comment in
-
Cell cycle: Continuous chromatin changes.Nature. 2017 Jul 5;547(7661):34-35. doi: 10.1038/547034a. Nature. 2017. PMID: 28682328 No abstract available.
-
Genome organization: Tracking chromosomal conformation through the cell cycle.Nat Rev Genet. 2017 Sep;18(9):514-515. doi: 10.1038/nrg.2017.62. Epub 2017 Jul 24. Nat Rev Genet. 2017. PMID: 28736439 No abstract available.
-
Chromosome structure dynamics during the cell cycle: a structure to fit every phase.EMBO J. 2017 Sep 15;36(18):2661-2663. doi: 10.15252/embj.201798014. Epub 2017 Sep 4. EMBO J. 2017. PMID: 28871059 Free PMC article.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
