

Metabolic labeling in middle-down proteomics allows for investigation of the dynamics of the histone code
- PMID: 28683815
- PMCID: PMC5501349
- DOI: 10.1186/s13072-017-0139-z
Metabolic labeling in middle-down proteomics allows for investigation of the dynamics of the histone code
Abstract
Background: Middle-down mass spectrometry (MS), i.e., analysis of long (~50-60 aa) polypeptides, has become the method with the highest throughput and accuracy for the characterization of combinatorial histone posttranslational modifications (PTMs). The discovery of histone readers with multiple domains, and overall the cross talk of PTMs that decorate histone proteins, has revealed that histone marks have synergistic roles in modulating enzyme recruitment and subsequent chromatin activities. Here, we demonstrate that the middle-down MS strategy can be combined with metabolic labeling for enhanced quantification of histone proteins and their combinatorial PTMs in a dynamic manner.
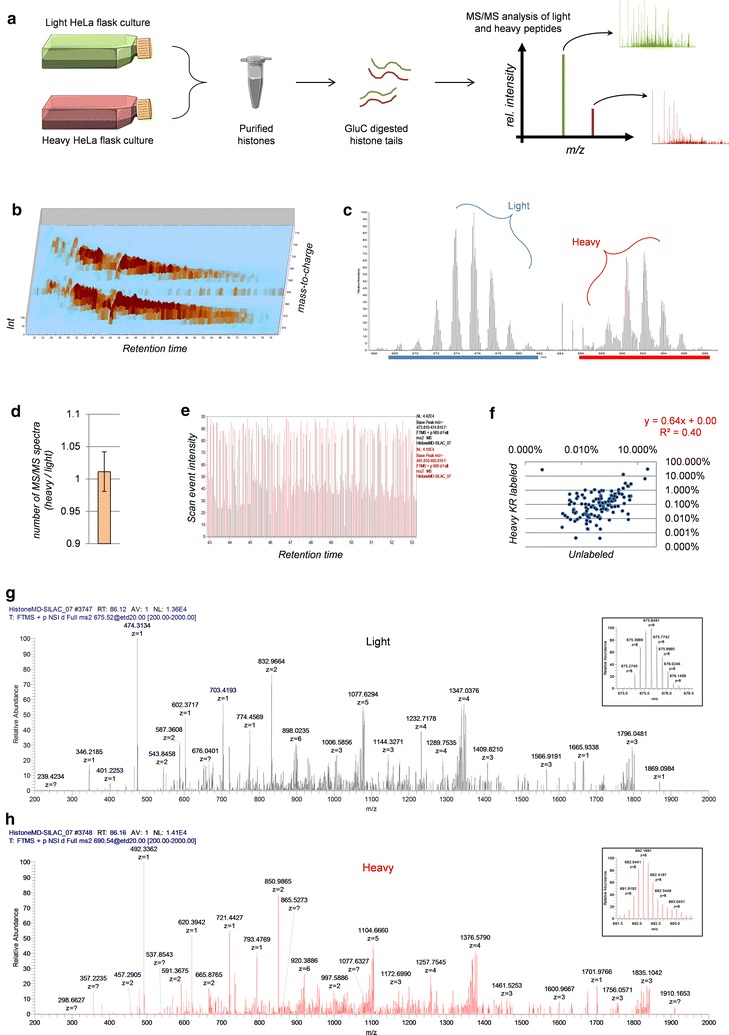
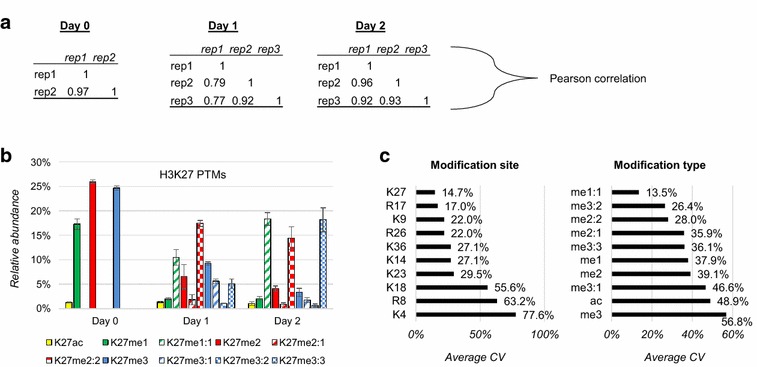
Methods: We used a nanoHPLC-MS/MS system consisting of hybrid weak cation exchange-hydrophilic interaction chromatography combined with high resolution MS and MS/MS with ETD fragmentation. After spectra identification, we filtered confident hits and quantified polypeptides using our in-house software isoScale.
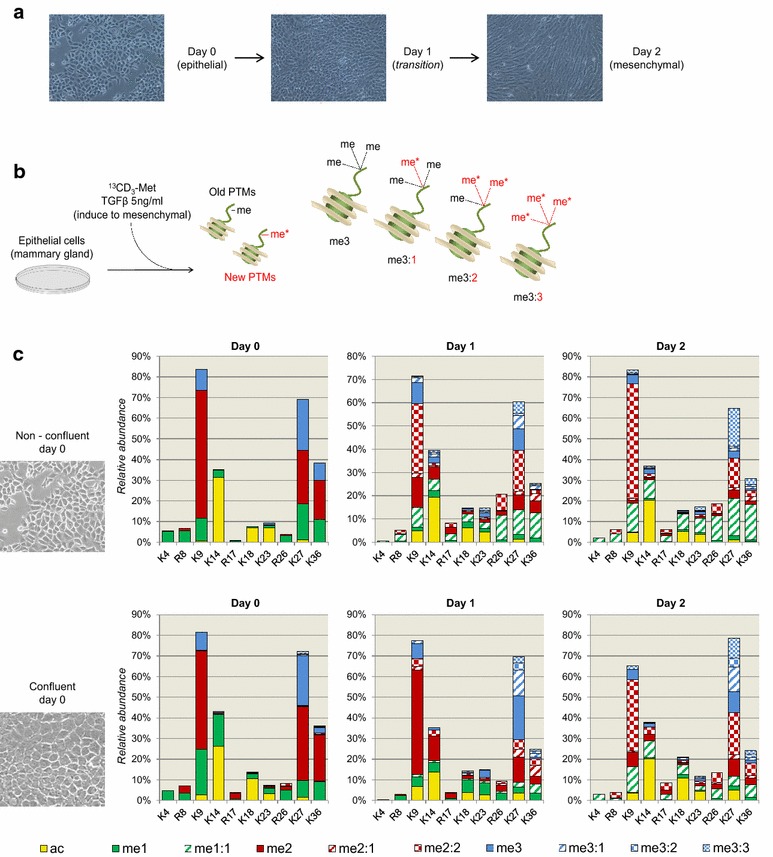
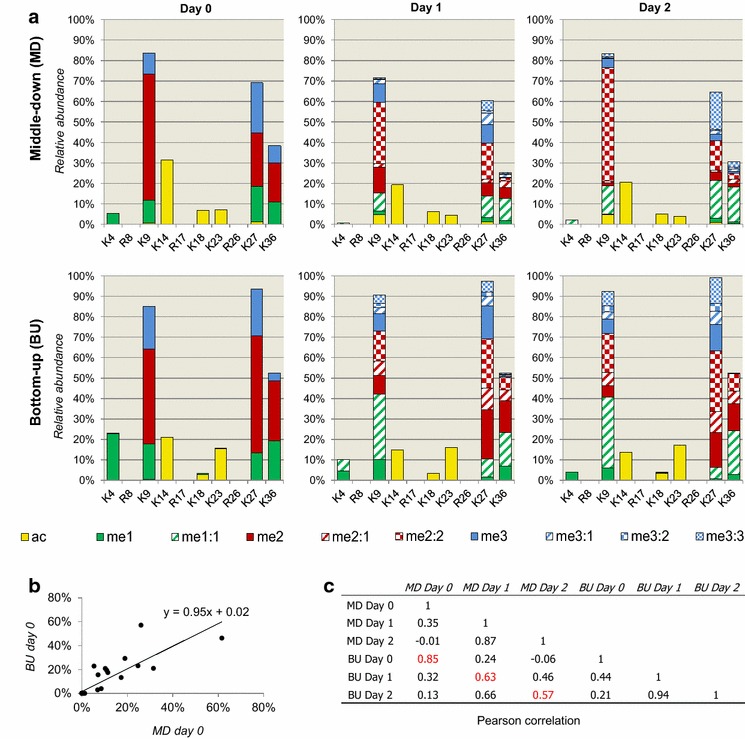
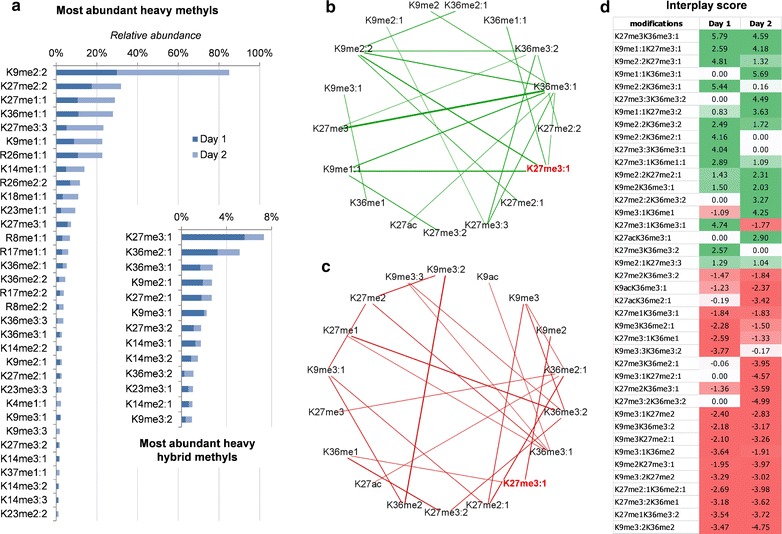
Results: We first verified that middle-down MS can discriminate and differentially quantify unlabeled from heavy labeled histone N-terminal tails (heavy lysine and arginine residues). Results revealed no bias toward identifying and quantifying unlabeled versus heavy labeled tails, even if the heavy labeled peptides presented the typical skewed isotopic pattern typical of long protein sequences that hardly get 100% labeling. Next, we plated epithelial cells into a media with heavy methionine-(methyl-13CD3), the precursor of the methyl donor S-adenosylmethionine and stimulated epithelial to mesenchymal transition (EMT). We assessed that results were reproducible across biological replicates and with data obtained using the more widely adopted bottom-up MS strategy, i.e., analysis of short tryptic peptides. We found remarkable differences in the incorporation rate of methylations in non-confluent cells versus confluent cells. Moreover, we showed that H3K27me3 was a critical player during the EMT process, as a consistent portion of histones modified as H3K27me2K36me2 in epithelial cells were converted into H3K27me3K36me2 in mesenchymal cells.
Conclusions: We demonstrate that middle-down MS, despite being a more scarcely exploited MS technique than bottom-up, is a robust quantitative method for histone PTM characterization. In particular, middle-down MS combined with metabolic labeling is currently the only methodology available for investigating turnover of combinatorial histone PTMs in dynamic systems.
Keywords: Epigenetics; Histones; Mass spectrometry; Methylation; Middle-down; Posttranslational modifications; SILAC.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
