

Arabidopsis ATRX Modulates H3.3 Occupancy and Fine-Tunes Gene Expression
- PMID: 28684426
- PMCID: PMC5559740
- DOI: 10.1105/tpc.16.00877
Arabidopsis ATRX Modulates H3.3 Occupancy and Fine-Tunes Gene Expression
Abstract
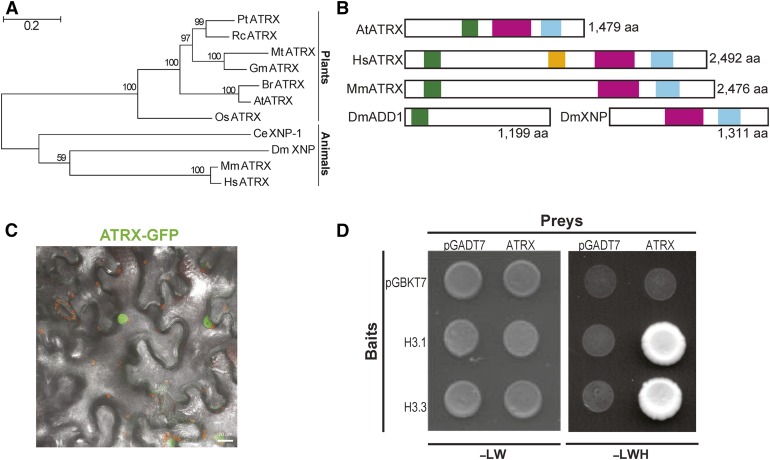
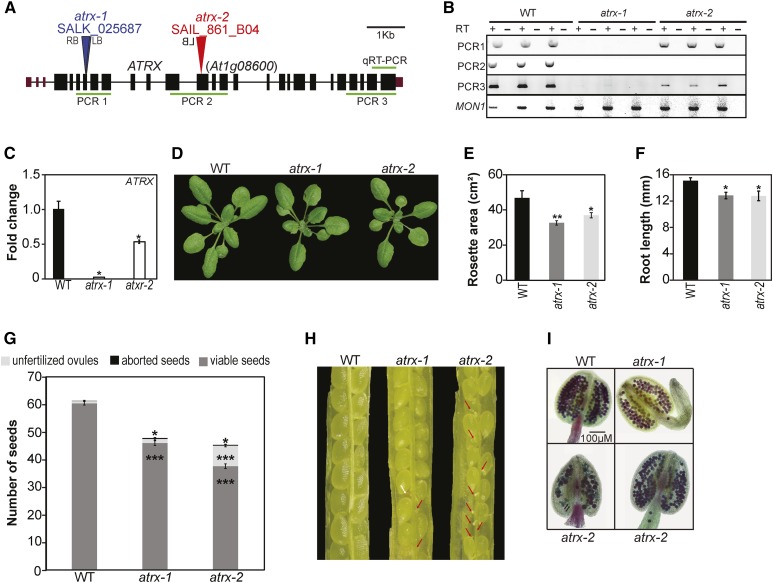
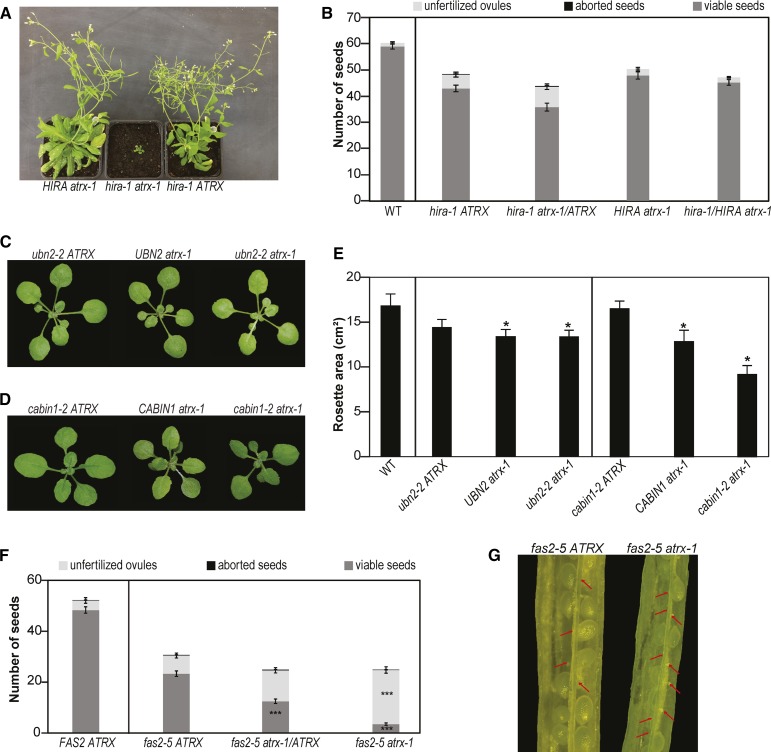
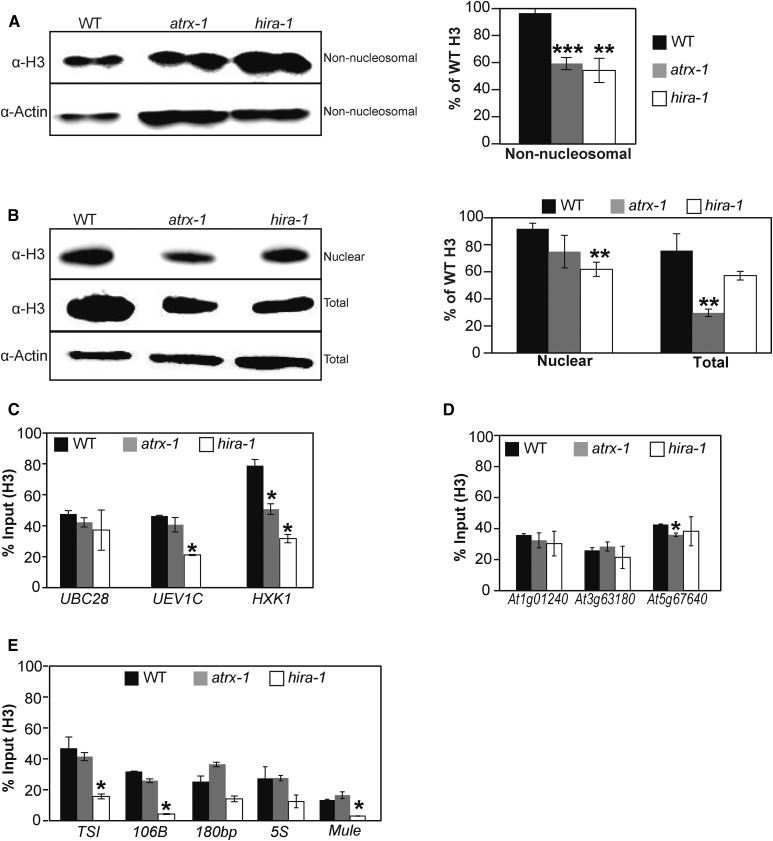
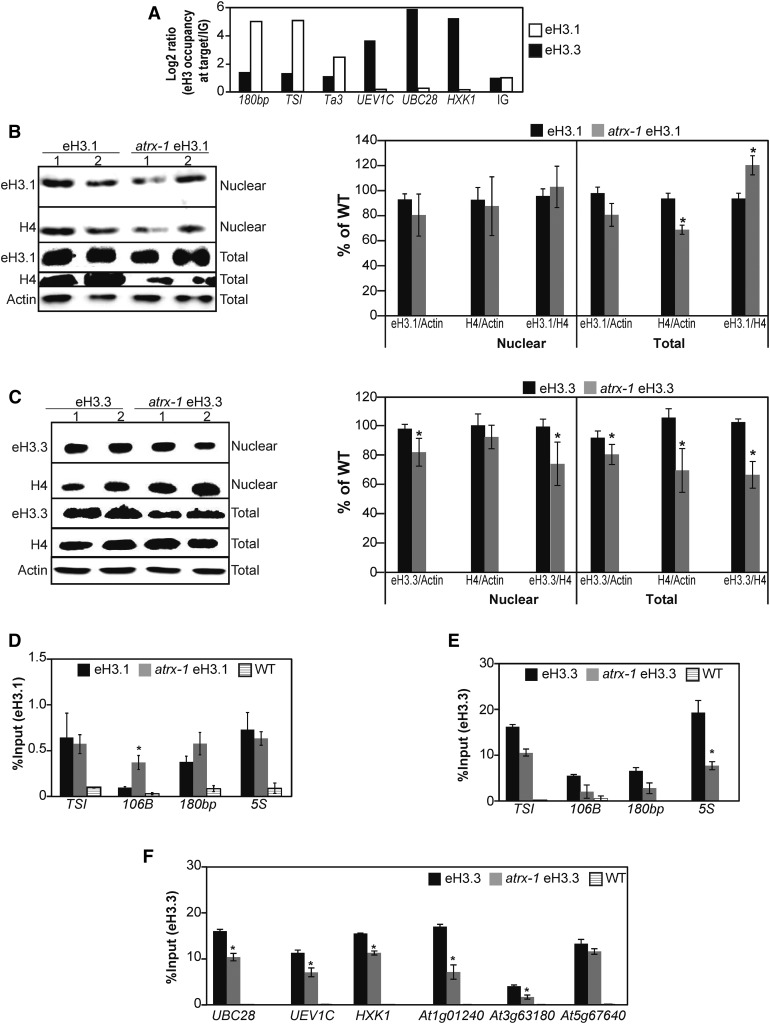
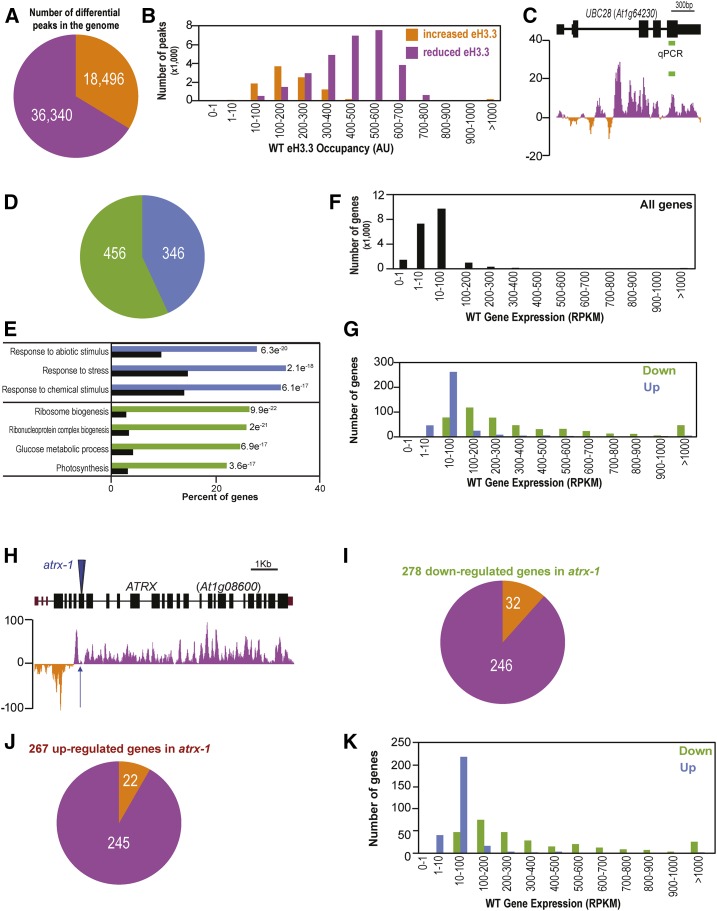
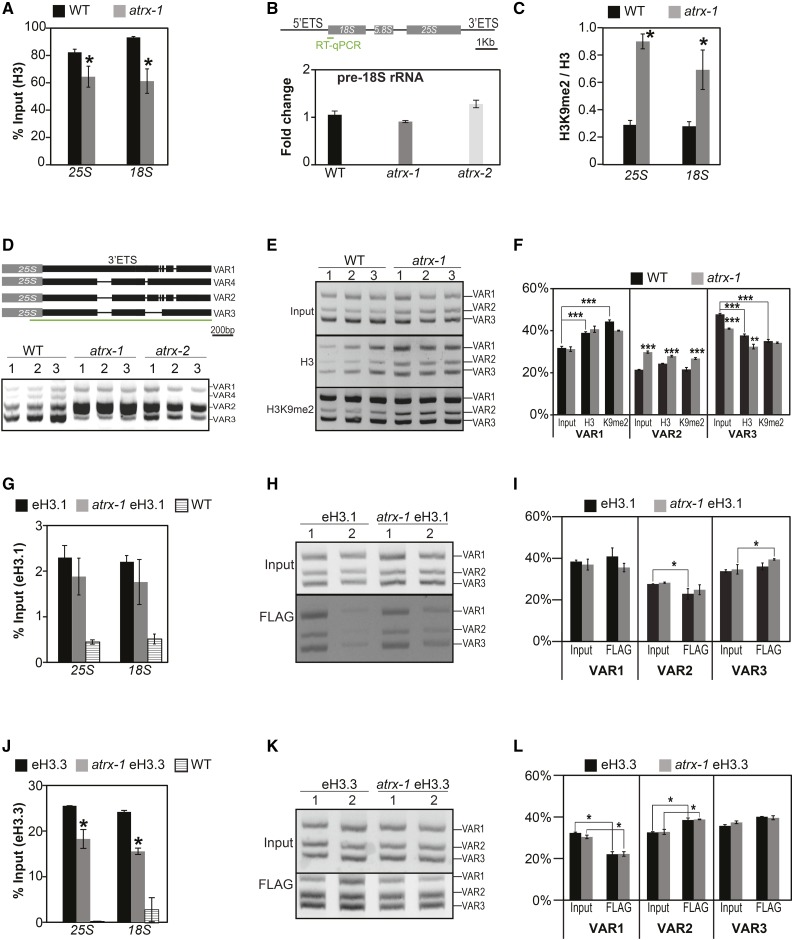
Histones are essential components of the nucleosome, the major chromatin subunit that structures linear DNA molecules and regulates access of other proteins to DNA. Specific histone chaperone complexes control the correct deposition of canonical histones and their variants to modulate nucleosome structure and stability. In this study, we characterize the Arabidopsis thaliana Alpha Thalassemia-mental Retardation X-linked (ATRX) ortholog and show that ATRX is involved in histone H3 deposition. Arabidopsis ATRX mutant alleles are viable, but show developmental defects and reduced fertility. Their combination with mutants of the histone H3.3 chaperone HIRA (Histone Regulator A) results in impaired plant survival, suggesting that HIRA and ATRX function in complementary histone deposition pathways. Indeed, ATRX loss of function alters cellular histone H3.3 pools and in consequence modulates the H3.1/H3.3 balance in the cell. H3.3 levels are affected especially at genes characterized by elevated H3.3 occupancy, including the 45S ribosomal DNA (45S rDNA) loci, where loss of ATRX results in altered expression of specific 45S rDNA sequence variants. At the genome-wide scale, our data indicate that ATRX modifies gene expression concomitantly to H3.3 deposition at a set of genes characterized both by elevated H3.3 occupancy and high expression. Together, our results show that ATRX is involved in H3.3 deposition and emphasize the role of histone chaperones in adjusting genome expression.
© 2017 American Society of Plant Biologists. All rights reserved.
Figures
References
-
- Abou-Ellail M., Cooke R., Sáez-Vásquez J. (2011). Variations in a team: major and minor variants of Arabidopsis thaliana rDNA genes. Nucleus 2: 294–299. - PubMed
-
- Alexander M.P. (1969). Differential staining of aborted and nonaborted pollen. Stain Technol. 44: 117–122. - PubMed
-
- Alexeev A., Mazin A., Kowalczykowski S.C. (2003). Rad54 protein possesses chromatin-remodeling activity stimulated by the Rad51-ssDNA nucleoprotein filament. Nat. Struct. Biol. 10: 182–186. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
