

PPR polyadenylation factor defines mitochondrial mRNA identity and stability in trypanosomes
- PMID: 28684539
- PMCID: PMC5556272
- DOI: 10.15252/embj.201796808
PPR polyadenylation factor defines mitochondrial mRNA identity and stability in trypanosomes
Abstract
In Trypanosoma brucei, most mitochondrial mRNAs undergo internal changes by RNA editing and 3' end modifications. The temporally separated and functionally distinct modifications are manifested by adenylation prior to editing, and by post-editing extension of a short A-tail into a long A/U-heteropolymer. The A-tail stabilizes partially and fully edited mRNAs, while the A/U-tail enables mRNA binding to the ribosome. Here, we identify an essential pentatricopeptide repeat-containing RNA binding protein, kinetoplast polyadenylation factor 3 (KPAF3), and demonstrate its role in protecting pre-mRNA against degradation by the processome. We show that KPAF3 recruits KPAP1 poly(A) polymerase to the 3' terminus, thus leading to pre-mRNA stabilization, or decay depending on the occurrence and extent of editing. In vitro, KPAF3 stimulates KPAP1 activity and inhibits mRNA uridylation by RET1 TUTase. Our findings indicate that KPAF3 selectively directs pre-mRNA toward adenylation rather than uridylation, which is a default post-trimming modification characteristic of ribosomal and guide RNAs. As a quality control mechanism, KPAF3 binding ensures that mRNAs entering the editing pathway are adenylated and, therefore, competent for post-editing A/U-tailing and translational activation.
Keywords: Trypanosoma; RNA editing; RNA stability; pentatricopeptide repeat; polyadenylation.
© 2017 The Authors.
Figures
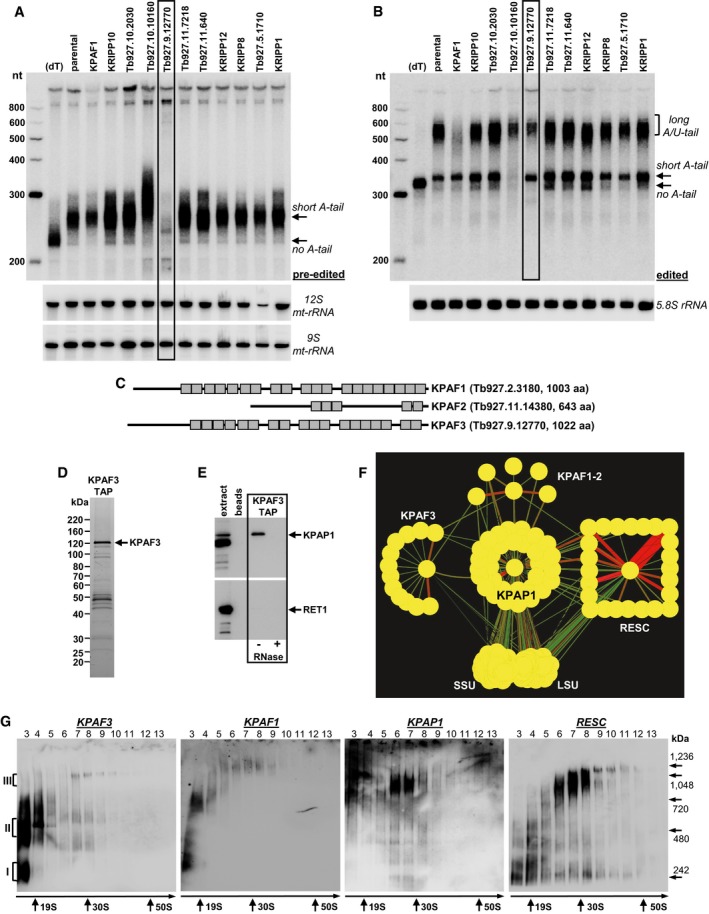
Representative Northern blotting of pre‐edited RPS12 mRNA and 9S and 12S mt‐rRNAs from targeted RNAi screen of predicted PPR proteins. Knockdown was induced for 72 h, and total RNA was separated on 5% polyacrylamide/8 M urea gel. Gene ID numbers are from
http://tritrypdb.org . (dT); RNA was hybridized with 20‐mer oligo(dT) and treated with RNase H to eliminate A‐tails. nt, RNA length in nucleotides. Positions of short A‐tailed and non‐adenylated transcripts are indicated by arrows.Same membrane as in (A) was hybridized with probes specific for edited RPS12 mRNA and cytoplasmic 5.8S rRNA (loading control). Edited RNAs with long A/U‐tail, short A‐tail, and non‐adenylated form are indicated.
Schematic repeat organization of kinetoplast polyadenylation factors 1, 2, and 3.
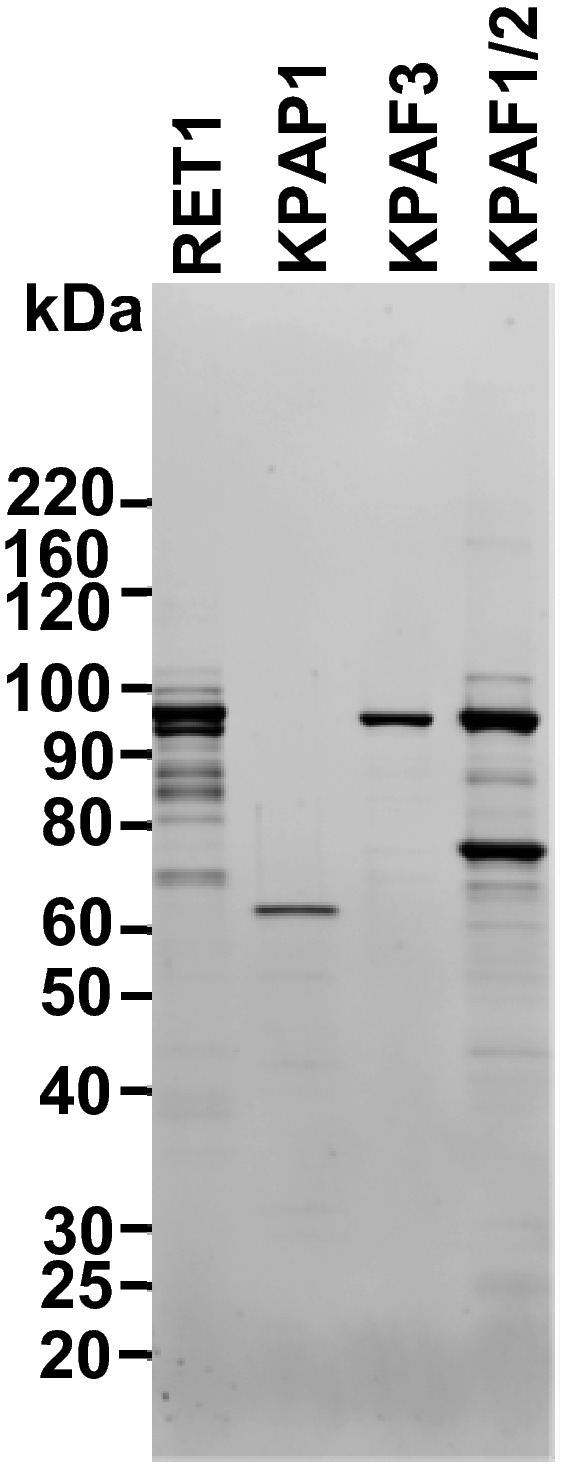
Tandem affinity purification of KPAF3. Final fraction was separated on 8–16% SDS gel and stained with Sypro Ruby.
KPAF3 fractions purified from mock‐ and RNase‐treated mitochondrial extracts were subjected to immunoblotting with antibodies against KPAP1 poly(A) polymerase and RET1 TUTase. Beads, purification from parental cell line.
Model of interactions between KPAP1 poly(A) polymerase, polyadenylation factors, RNA editing substrate binding complex (RESC), and the ribosome. KPAP1, KPAF2, KPAF3, GRBC1, L3, and S17 proteins were affinity purified from mitochondrial lysates. The network was generated in Cytoscape software from bait–prey pairs in which the prey protein was identified by five or more unique peptides (Table EV1). The edge thickness correlates with normalized spectral abundance factor (NSAF) values.
Mitochondrial fraction was extracted with detergent, and soluble contents were separated for 5 h at 178,000 g in 10–30% glycerol gradient. Each fraction was further resolved on 3–12% Bis‐Tris native gel. Positions of native protein standards are denoted by arrows. KPAP1 and KPAF3 were visualized by immunoblotting. RNA editing substrate binding complex (RESC) was detected with antibodies against guide RNA binding subunits GRBC1 and GRBC2. Thyroglobulin (19S) and bacterial ribosomal subunits were used as apparent S‐value standards.
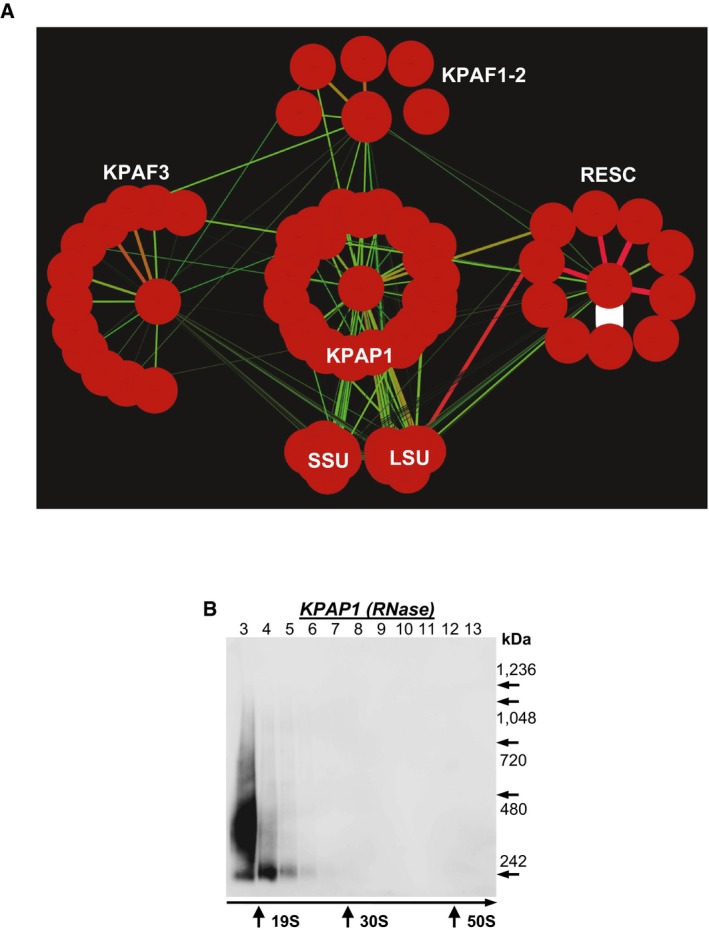
KPAP1, KPAF2, KPAF3, GRBC1, L3, and S17 proteins were affinity purified from mitochondrial lysates in the presence of RNase A (0.1 mg/ml) and RNase T1 (500 U/ml). The network was generated in Cytoscape software from bait–prey pairs in which the prey protein was identified by five or more unique peptides (Table EV1). The edge thickness correlates with NSAF values.
Mitochondrial fraction was extracted with detergent in the presence of RNase A (0.1 mg/ml) and RNase T1 (500 U/ml), and soluble contents were separated for 5 h at 178,000 g in 10–30% glycerol gradient. Each fraction was further resolved on 3–12% Bis‐Tris native gel. Positions of native protein standards are denoted by arrows. KPAP1 was visualized by immunoblotting. Thyroglobulin (19S) and bacterial ribosomal subunits were used as apparent S‐value standards.
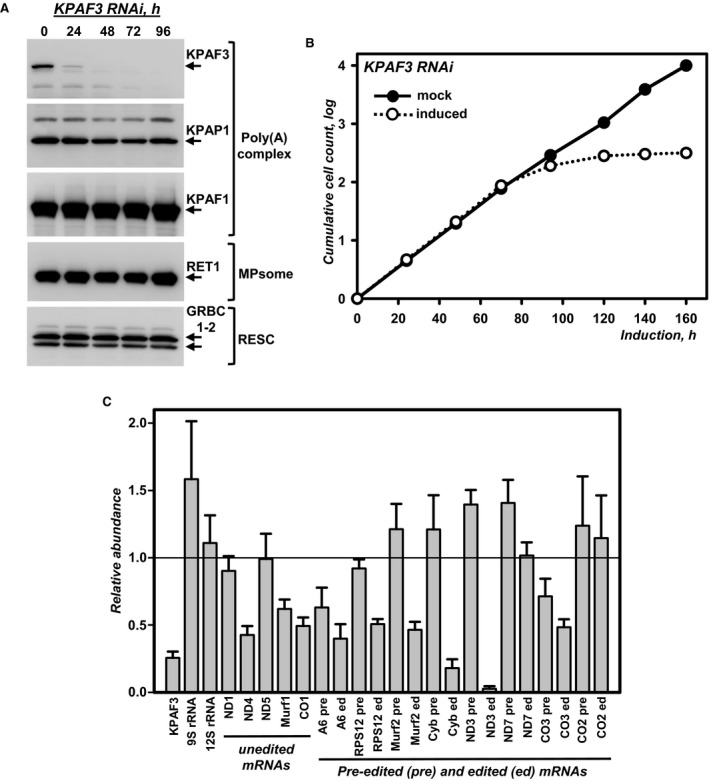
Cell lysates prepared at indicated time points of KPAF3 RNAi induction were subjected to sequential immunoblotting with antigen‐purified antibodies against KPAF3, KPAP1, KPAF1, GRBC1‐2, and monoclonal antibodies against RET1 TUTase.
Growth kinetics of procyclic parasite suspension cultures after mock induction and RNAi expression.
Quantitative real‐time RT–PCR analysis of RNAi‐targeted KPAF3 mRNA, and mitochondrial rRNAs and mRNAs. The assay distinguishes edited and corresponding pre‐edited transcripts, and unedited mRNAs. RNA levels were normalized to β‐tubulin mRNA. RNAi was induced for 55 h. Error bars represent the standard deviation from at least three replicates. The thick line at “1” reflects no change in relative abundance; bars above or below represent an increase or decrease, respectively. Pre, pre‐edited mRNA; ed, edited mRNA.
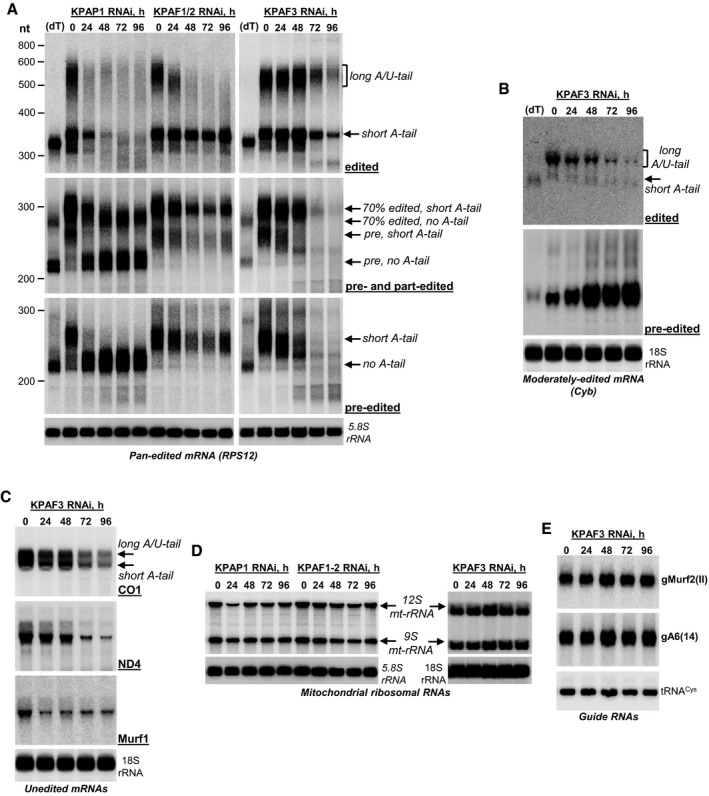
Northern blotting of pre‐edited, partially edited, and fully edited RPS12 mRNAs. Total RNA was separated on a 5% polyacrylamide/8 M urea gel and sequentially hybridized with radiolabeled single‐stranded DNA probes. Zero time point refers to mock‐induced RNAi cell line. Cytosolic ribosomal RNA (5.8S) was used as loading control. (dT), RNA was hybridized with 20‐mer oligo(dT) and treated with RNase H to eliminate A‐tails. Positions of short A‐tailed and non‐adenylated transcripts are indicated by arrows. Pre, pre‐edited mRNA. Partially edited mRNAs (70% edited) were detected with a probe complementary to positions 33–70 in pre‐edited RPS12 mRNA.
Northern blotting of moderately edited cyb mRNA. Total RNA was separated on a 1.7% agarose/formaldehyde gel and sequentially hybridized with oligonucleotide probes selective for pre‐edited and fully edited sequences. Cytosolic 18S ribosomal RNA was used as loading control.
Northern blotting of unedited mRNA. Total RNA was separated on a 1.7% agarose/formaldehyde gel and hybridized with oligonucleotide probes. Cytosolic 18S rRNA was used as loading control.
Northern blotting of mitochondrial ribosomal RNAs. Total RNA was separated on a 5% polyacrylamide/8 M urea (KPAP1 and KPAF1‐2 RNAi) or 1.8% agarose (KPAF3 RNAi) gels and hybridized with radiolabeled single‐stranded DNA probes specific for 9S and 12S mt‐rRNA. Cytosolic ribosomal RNAs were used as loading controls.
Guide RNA Northern blotting. Total RNA was separated on a 10% polyacrylamide/8 M urea gel and hybridized with oligonucleotide probes specific for maxicircle‐encoded gMurf2(II) and minicircle‐encoded gA6(14). Mitochondrially localized tRNACys was used as loading control.
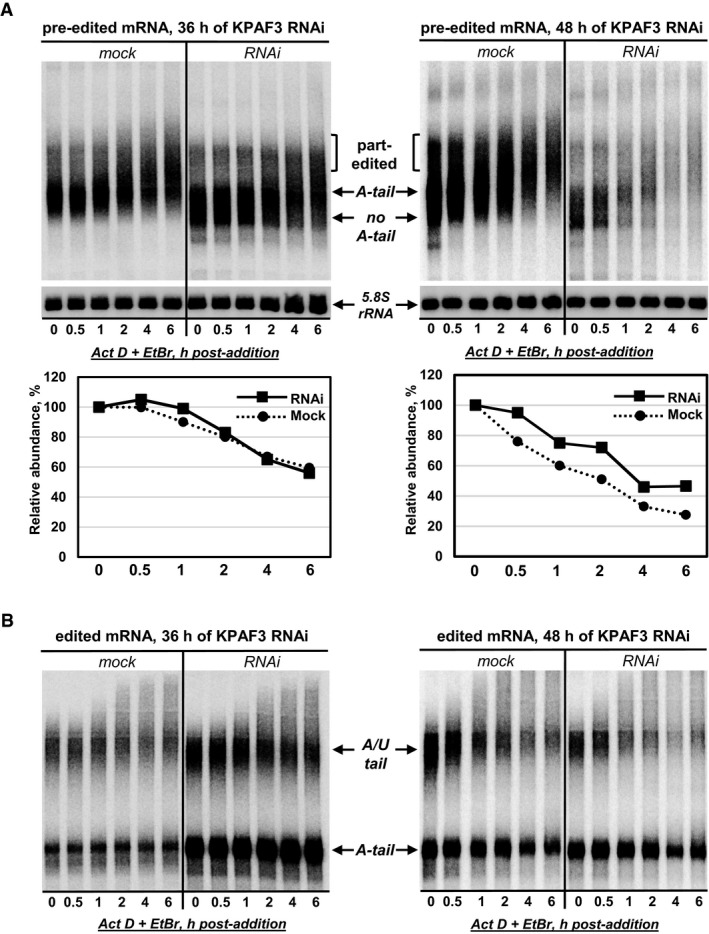
Pre‐edited mRNA decay in KPAF3 RNAi cells at 36 h (left panel) and 48 h (right panel) post‐induction. After RNAi induction, Actinomycin D and ethidium bromide were added to inhibit transcription. Total RNA was isolated from cells collected at indicated time points after ActD/EtBr bromide addition, separated on denaturing 5% PAGE, and hybridized with DNA probe for pre‐edited RPS12 mRNA. Quantitation was performed in reference to 5.8S rRNA. The graphs below Northern blotting panels represent changes in relative abundance, assuming the mRNA/5.8S rRNA ratio at the time of ActD/EtBr addition as 100%. Contrast was increased in the right panel to reflect RNA loss at 48 h of KPAF3 RNAi.
Fully edited RPS12 mRNA decay in KPAF3 RNAi cells. Same membrane as in (A) was hybridized with a probe specific for a fully edited RPS12 mRNA.

KPAP1 and RET1 were incubated with 5′‐labeled 66‐mer RNA, ATP, and UTP, as indicated, in the presence of increasing KPAF3 amounts. Protein inclusion in combinatorial reactions is indicated by brackets.
KPAP1 was incubated with 5′‐labeled 66‐mer RNA and ATP in the presence of increasing KPAF1‐2 and KPAF3 amounts.
RET1 was incubated with 5′‐labeled 66‐mer RNA and UTP in the presence of increasing KPAF1‐2 and KPAF3 amounts for 20 min.
KPAP1 was incubated with 5′‐labeled 24‐mer RNAs and ATP in the presence of increasing KPAF3 amounts.
KPAP1 was incubated with 5′‐labeled 24‐mer [6A] RNA, ATP, and increasing concentrations of indicated homopolymers in the absence (left panel) or presence (right panel) of fixed KPAF3 amount.
KPAP1 was incubated with 5′‐labeled 24‐mer RNA terminating with 6As, various NTPs, and fixed KPAF3 amount, as indicated.
- A
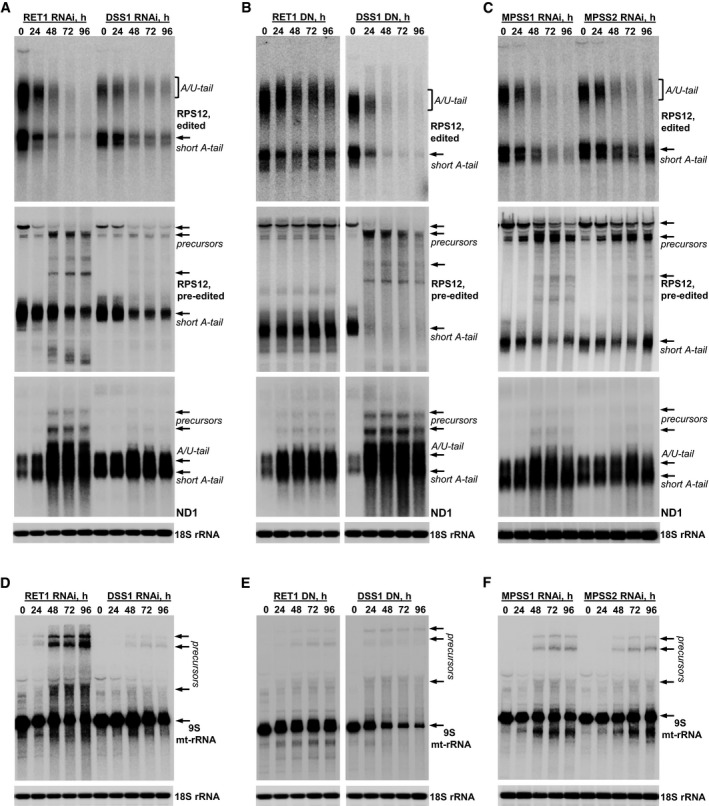
Edited and pre‐edited RPS12 mRNA, and unedited ND1 mRNA were analyzed in RET1 and DSS1 RNAi cells. Precursors are indicated by arrows.
- B
Edited and pre‐edited RPS12 mRNA, and unedited ND1 mRNA were analyzed in cell lines overexpressing dominant negative (DN) RET1 and DSS1 variants.
- C
Edited and pre‐edited RPS12 mRNA, and unedited ND1 mRNA were analyzed in MPSS1 and MPSS2 RNAi cells.
- D–F
Same membranes as shown in panels (A–C) were hybridized with radiolabeled oligonucleotide probes specific for 9S mt‐rRNA.
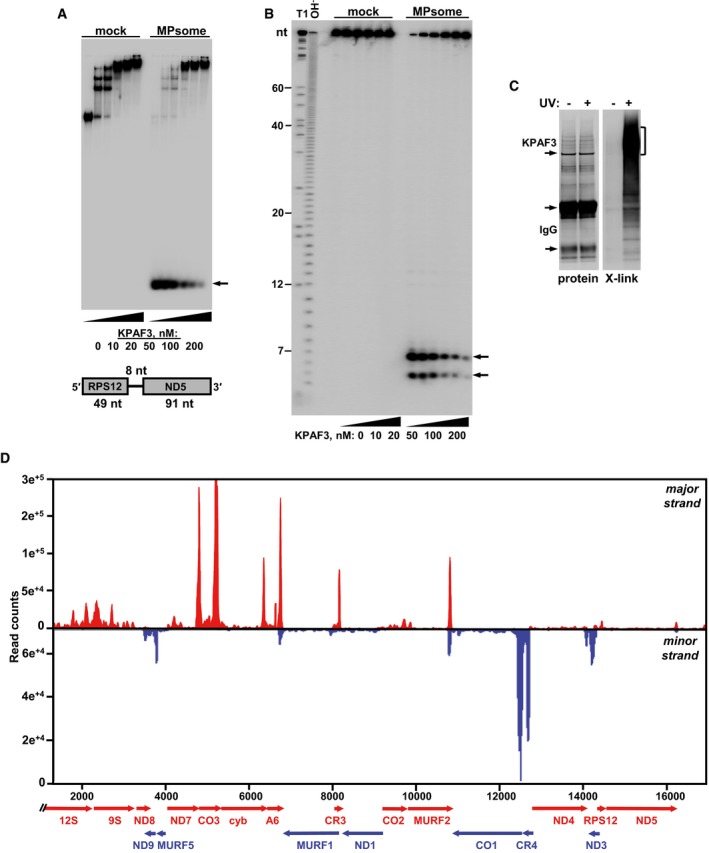
Electrophoretic mobility shift assay. Increasing amounts of recombinant KPAF3 were incubated with 5′‐radiolabeled RNA in the absence (mock) or presence (MPsome) of tandem affinity‐purified DSS1 exonuclease. Reactions were separated on 7% Tris‐borate native PAGE. Degradation products are indicated by arrows.
Analysis of RNA degradation patterns. Same reaction products as in (A) were deproteinized and separated on high‐resolution 15% polyacrylamide/8 M urea gel. T1, RNA was digested with G‐specific RNase T1. OH−, alkaline ladder.
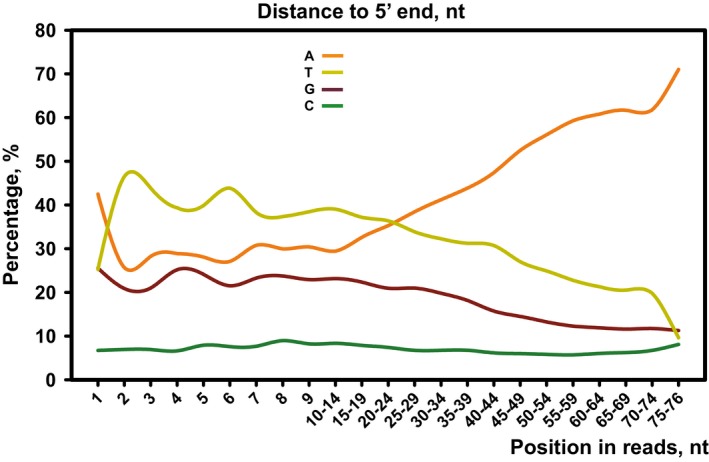
Isolation of in vivo RNA‐KPAF3 crosslinks. KPAF3‐TAP fusion protein was affinity purified from parasites subjected to UV‐irradiation (+) or mock‐treated (−). Final fractions were subjected to partial RNase I digestion, and RNA fragments covalently bound to the protein were radiolabeled. Upon separation on SDS–PAGE, RNA‐protein crosslinks were transferred onto nitrocellulose membrane. Protein patterns were visualized by Sypro Ruby staining (left panel) and RNA‐protein crosslinks by exposure to phosphor storage screen (right panel). RNA was eluted from areas indicated by brackets, converted into barcoded libraries, and sequenced.
In vivo positioning of KPAF3 binding sites. Crosslinked fragments were mapped to gene‐containing region of the maxicircle. Annotated mitochondrial transcripts encoded on major strand are indicated by red arrows, and those on minor strand are delineated by blue arrows.
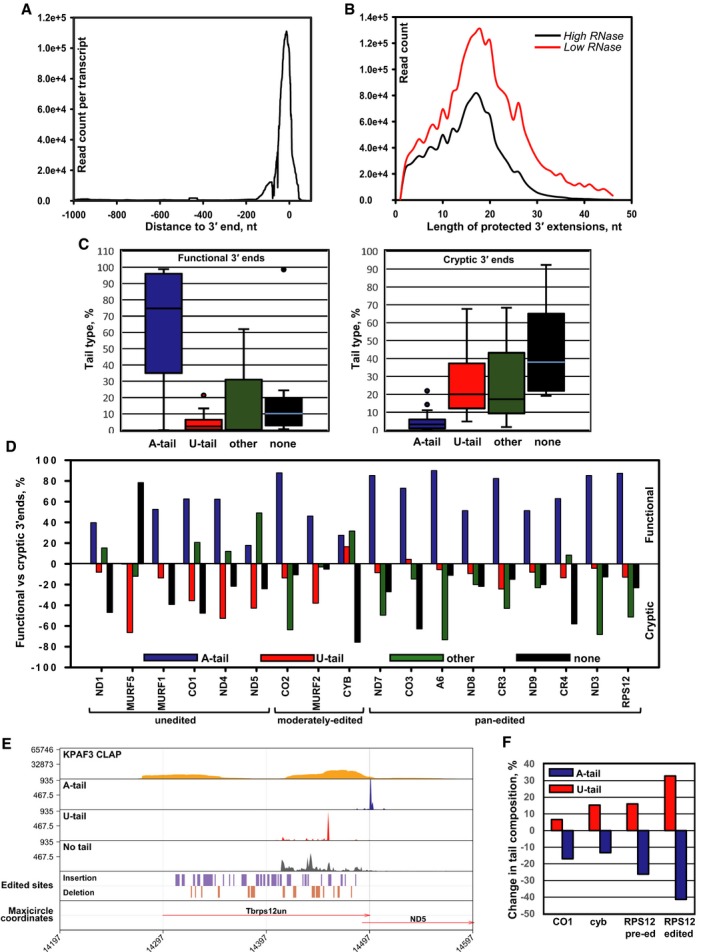
Overall distribution of KPAF3 binding sites in mitochondrial mRNAs. The CLAP‐Seq reads were aligned to unedited and fully edited sequences. Read counts located 1,000 nt upstream and 100 nt downstream of the 3′ end in each transcript were collected in 1 nt bins. The average coverage across all genes was plotted.
Length distribution of KPAF3‐protected extensions. The 3′ end sequences that do not align to encoded or edited 3′ ends were considered as 3′ modifications. The length distribution of KPAF3‐protected tail sequences was derived from KPAF3 CLAP‐Seq experiments performed at low and high RNase I concentrations.
Global modification status of functional and cryptic 3′ termini. Based on 3′ RACE data, the 3′ end patterns were classified as functional (left graph, include stop codon and 3′ UTR) and cryptic (right graph, truncations extend into coding sequence). The percentages of A‐tailed (> 90% As), U‐tailed (> 90% Us), unmodified (lack unmapped 3′ overhangs), and other (those that do not fall into previous three categories) were calculated for all mitochondrial mRNAs. The boxplot distribution of 3′ modification patterns for functional termini in all transcripts is shown on the left panel, and cryptic ends are plotted on the right panel. The horizontal line within each boxplot indicates the median value for all mRNAs. The error bars indicate the 1.5× interquartile range (IQR) distance from median. IQR is defined as the distance between the 25%–75% quantiles. The upper and lower bounds of the box indicate the 75% and 25% quantiles.
Transcript‐specific differences in modification patterns between functional (upper panel) and cryptic (lower panel) 3′ termini.
KPAF3 binding correlates with downstream adenylation events. A‐tailed, U‐tailed, and unmodified 3′ ends of pre‐edited RPS12 mRNAs were mapped along with KPAF3 CLAP‐Seq reads and positions of editing events onto maxicircle DNA. Gene positions are shown by arrows.
Representative unedited (CO1), moderately edited (cyb), and pan‐edited (RPS12) mRNAs were subjected to 3′ RACE in mock‐induced and KPAF3 RNAi knockdown cell line. The percentage of different tail types was calculated for the pre‐edited or edited form (not distinguished in 5′ edited cyb mRNA). The changes in A‐tail and U‐tail percentage at the functional 3′ ends were compared between control and KPAF3 knockdown cells.
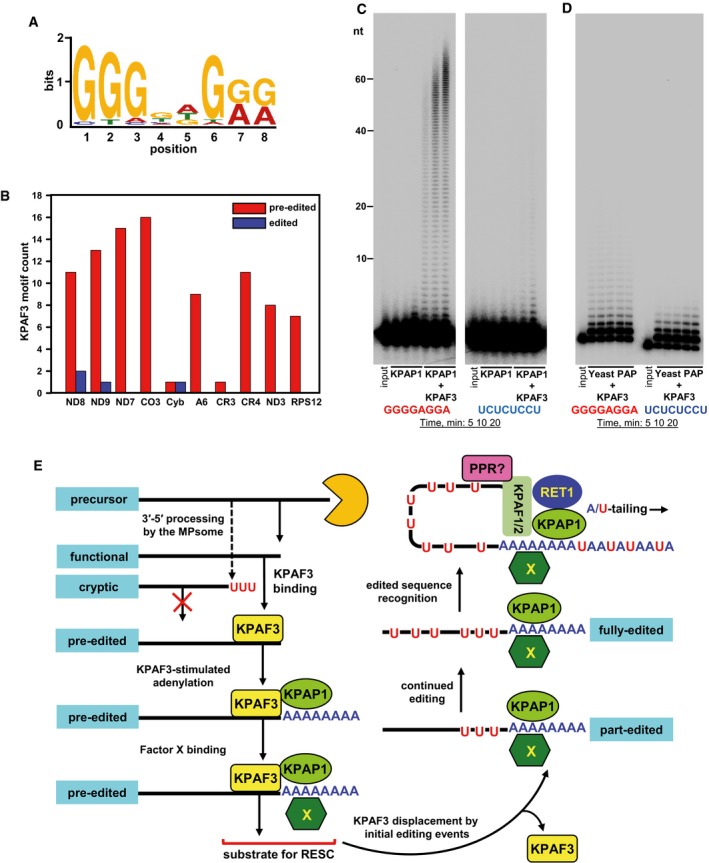
KPAF3 in vivo binding motif. The MACS algorithm was used to call KPAF3 CLAP‐Seq peaks separately on both maxicircle strands. The significant peaks from samples treated with low and high RNase I concentrations were extended on both sides by 100 nt and used as input; the maxicircle sequences were used as the background model. The MEME algorithm was applied to predict the enriched motif for KPAF3 binding.
Distribution of KPAF3 binding sites between pre‐edited and edited transcripts. The motif shown in panel (A) was queried against edited and pre‐edited maxicircle transcripts using FIMO algorithm with a P‐value cutoff at 0.001. The number of predicted motifs in pre‐edited and edited maxicircle transcripts was plotted as a bar graph.
Motif‐dependent stimulation of KPAP1 poly(A) polymerase by KPAF3. Synthetic 5′‐radiolabeled 40‐mers containing either predicted G‐rich motif (left panel), or arbitrary pyrimidine octamer (right panel), was incubated with 100 nM of KPAP1 in the presence or absence of 100 nM of KPAF3. Reactions were performed for 5, 10, 20 min, and products were resolved on 10% polyacrylamide/8 M urea gel.
Specificity of KPAP1 poly(A) polymerase stimulation by KPAF3. The assay was performed with 14 nM of yeast poly(A) polymerase in the presence of 0, 25, 50, 100, and 200 nM of KPAF3 for 20 min, and products were resolved on 10% polyacrylamide/8 M urea gel.
Model for functional coupling of primary precursor processing, adenylation, and editing processes. The MPsome‐catalyzed 3′–5′ degradation pauses near the mature 3′ end by a still‐unknown mechanism. Upon pausing, however, two outcomes become feasible depending on the KPAF3 binding site's proximity to the 3′ end: (i) KPAF3 recruits KPAP1 poly(A) polymerase and stimulates short A‐tail addition to downstream terminus; and (ii) lack of bound KPAF3 causes MPsome to dissociate leaving either the unmodified 3′ end, or that with RET1‐added U‐tail. The former modification likely designates the transcript as mRNA, while the latter occurs on rRNAs and truncated mRNA species. A hypothetical factor X is proposed to bind the A‐tail to stabilize edited mRNA once the editing machinery displaces KPAF3 from the 3′ region. Addition of long A/U‐tail to a pre‐existing 3′ A‐tail is triggered upon completion of editing, which typically occurs at the 5′ end. Hence, we hypothesize the existence of a PPR factor that recognizes the RNA sequence created de novo by editing, and recruits KPAF1/2 factors and RET1 TUTase to short A‐tail preloaded with KPAP1 and Factor X. This event likely triggers A/U‐tailing, leading to translational activation (Aphasizheva et al, 2011).
Similar articles
-
Mitochondrial RNA quality control in trypanosomes.Wiley Interdiscip Rev RNA. 2021 May;12(3):e1638. doi: 10.1002/wrna.1638. Epub 2020 Dec 16. Wiley Interdiscip Rev RNA. 2021. PMID: 33331073 Free PMC article. Review.
-
Poly(A) binding KPAF4/5 complex stabilizes kinetoplast mRNAs in Trypanosoma brucei.Nucleic Acids Res. 2020 Sep 4;48(15):8645-8662. doi: 10.1093/nar/gkaa575. Nucleic Acids Res. 2020. PMID: 32614436 Free PMC article.
-
Pentatricopeptide repeat proteins stimulate mRNA adenylation/uridylation to activate mitochondrial translation in trypanosomes.Mol Cell. 2011 Apr 8;42(1):106-17. doi: 10.1016/j.molcel.2011.02.021. Mol Cell. 2011. PMID: 21474072 Free PMC article.
-
3' adenylation determines mRNA abundance and monitors completion of RNA editing in T. brucei mitochondria.EMBO J. 2008 Jun 4;27(11):1596-608. doi: 10.1038/emboj.2008.87. Epub 2008 May 8. EMBO J. 2008. PMID: 18464794 Free PMC article.
-
Mitochondrial RNA editing in trypanosomes: small RNAs in control.Biochimie. 2014 May;100:125-31. doi: 10.1016/j.biochi.2014.01.003. Epub 2014 Jan 17. Biochimie. 2014. PMID: 24440637 Free PMC article. Review.
Cited by
-
Separating the Wheat from the Chaff: RNA Editing and Selection of Translatable mRNA in Trypanosome Mitochondria.Pathogens. 2019 Jul 18;8(3):105. doi: 10.3390/pathogens8030105. Pathogens. 2019. PMID: 31323762 Free PMC article. Review.
-
Pentatricopeptide repeat poly(A) binding protein KPAF4 stabilizes mitochondrial mRNAs in Trypanosoma brucei.Nat Commun. 2019 Jan 11;10(1):146. doi: 10.1038/s41467-018-08137-2. Nat Commun. 2019. PMID: 30635574 Free PMC article.
-
Mitochondrial RNA quality control in trypanosomes.Wiley Interdiscip Rev RNA. 2021 May;12(3):e1638. doi: 10.1002/wrna.1638. Epub 2020 Dec 16. Wiley Interdiscip Rev RNA. 2021. PMID: 33331073 Free PMC article. Review.
-
The 27 kDa Trypanosoma brucei Pentatricopeptide Repeat Protein is a G-tract Specific RNA Binding Protein.Sci Rep. 2018 Nov 19;8(1):16989. doi: 10.1038/s41598-018-34377-9. Sci Rep. 2018. PMID: 30451852 Free PMC article.
-
Manipulation of mitochondrial poly(A) polymerase family proteins in Trypanosoma brucei impacts mRNA termini processing.Front Parasitol. 2024 Jan 11;2:1298561. doi: 10.3389/fpara.2023.1298561. eCollection 2023. Front Parasitol. 2024. PMID: 39816830 Free PMC article.
References
-
- Aphasizhev R, Aphasizheva I (2007) RNA editing uridylyltransferases of trypanosomatids. Methods Enzymol 424: 51–67 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
