

Statistically robust methylation calling for whole-transcriptome bisulfite sequencing reveals distinct methylation patterns for mouse RNAs
- PMID: 28684555
- PMCID: PMC5580717
- DOI: 10.1101/gr.210666.116
Statistically robust methylation calling for whole-transcriptome bisulfite sequencing reveals distinct methylation patterns for mouse RNAs
Abstract
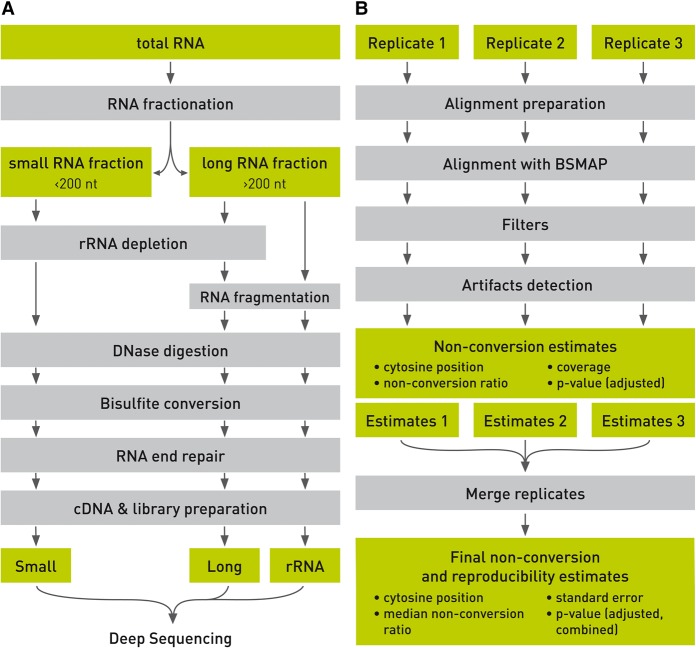
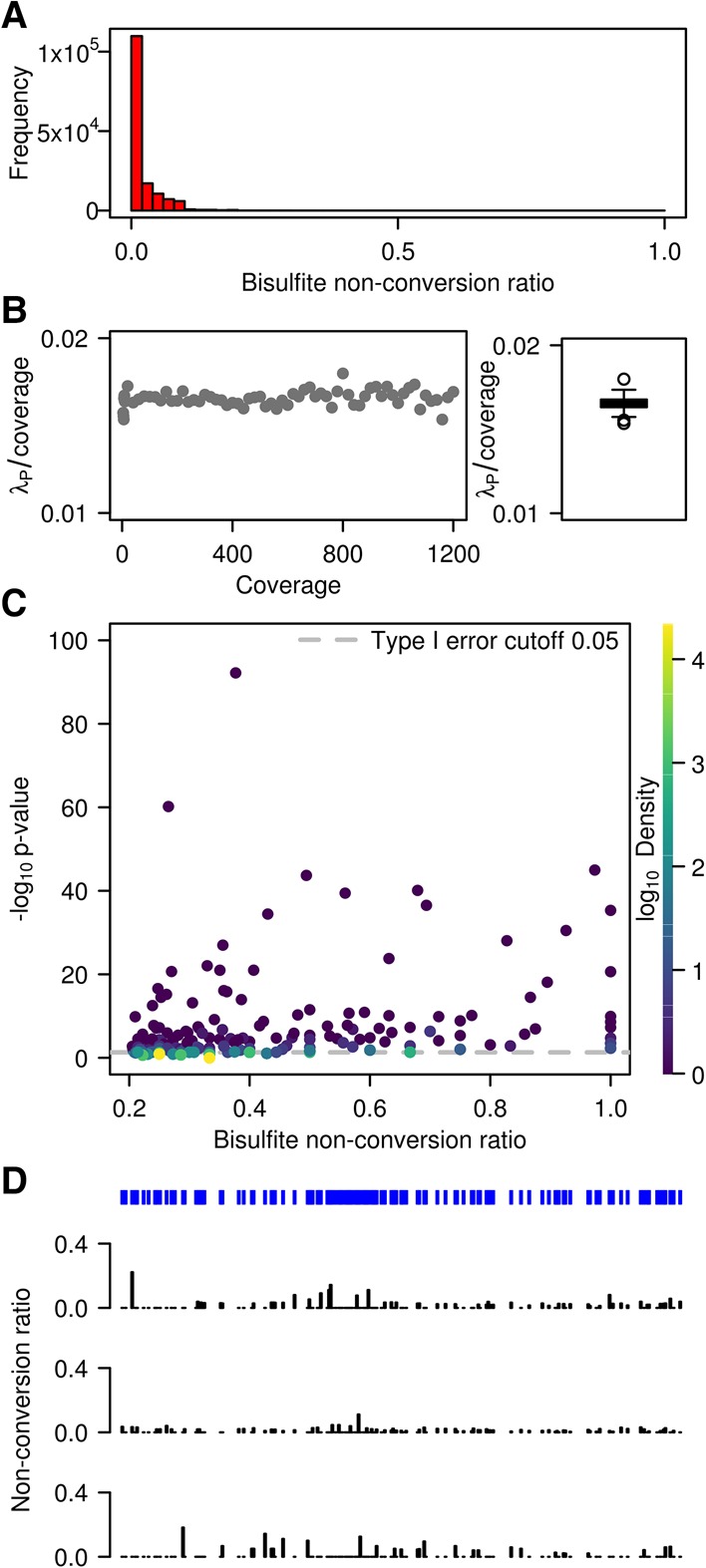
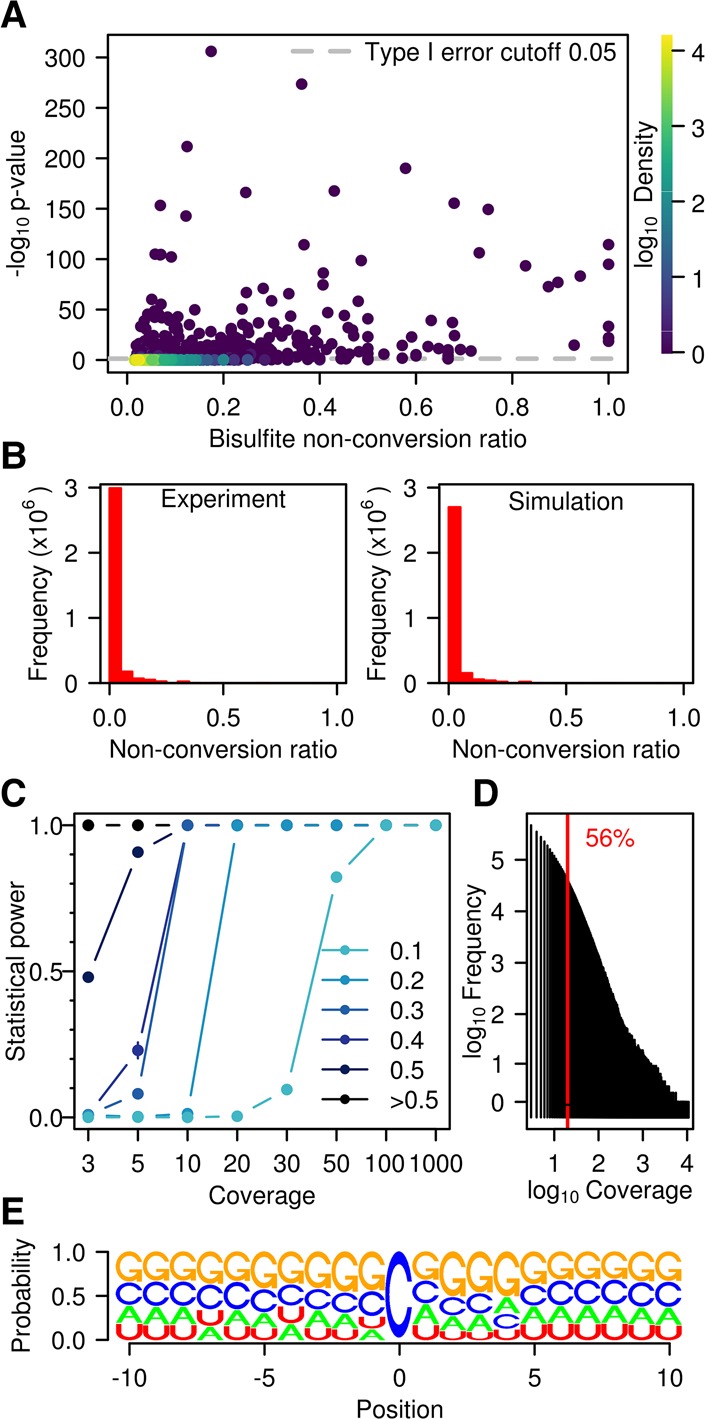
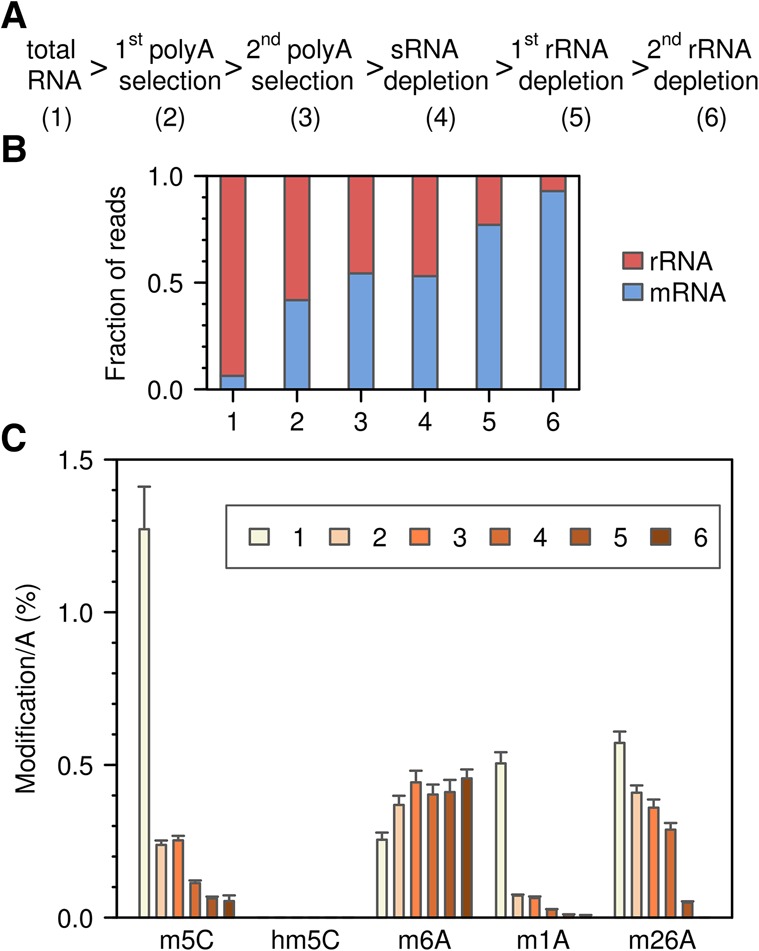
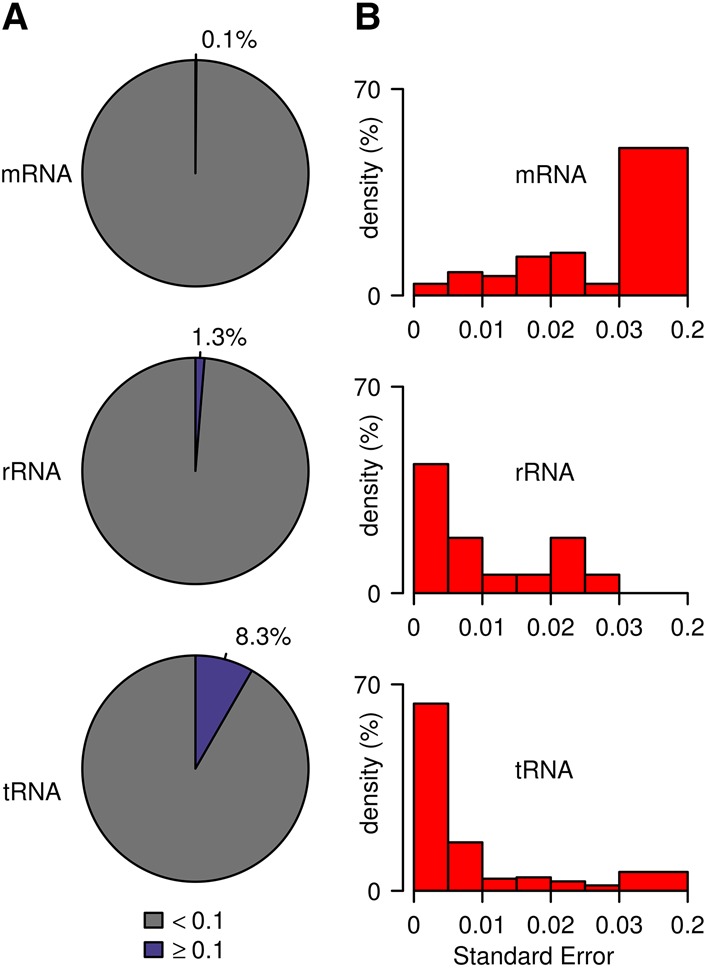
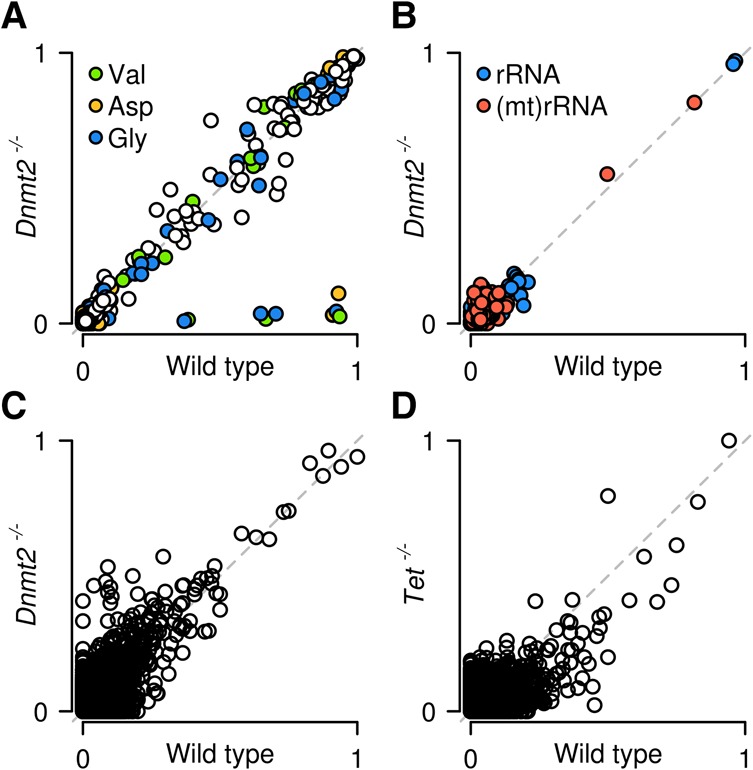
Cytosine-5 RNA methylation plays an important role in several biologically and pathologically relevant processes. However, owing to methodological limitations, the transcriptome-wide distribution of this mark has remained largely unknown. We previously established RNA bisulfite sequencing as a method for the analysis of RNA cytosine-5 methylation patterns at single-base resolution. More recently, next-generation sequencing has provided opportunities to establish transcriptome-wide maps of this modification. Here, we present a computational approach that integrates tailored filtering and data-driven statistical modeling to eliminate many of the artifacts that are known to be associated with bisulfite sequencing. By using RNAs from mouse embryonic stem cells, we performed a comprehensive methylation analysis of mouse tRNAs, rRNAs, and mRNAs. Our approach identified all known methylation marks in tRNA and two previously unknown but evolutionary conserved marks in 28S rRNA. In addition, mRNAs were found to be very sparsely methylated or not methylated at all. Finally, the tRNA-specific activity of the DNMT2 methyltransferase could be resolved at single-base resolution, which provided important further validation. Our approach can be used to profile cytosine-5 RNA methylation patterns in many experimental contexts and will be important for understanding the function of cytosine-5 RNA methylation in RNA biology and in human disease.
© 2017 Legrand et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
Similar articles
-
BisAMP: A web-based pipeline for targeted RNA cytosine-5 methylation analysis.Methods. 2019 Mar 1;156:121-127. doi: 10.1016/j.ymeth.2018.10.013. Epub 2018 Oct 24. Methods. 2019. PMID: 30366099
-
RNA 5-Methylcytosine Analysis by Bisulfite Sequencing.Methods Enzymol. 2015;560:297-329. doi: 10.1016/bs.mie.2015.03.007. Epub 2015 May 13. Methods Enzymol. 2015. PMID: 26253976
-
Conservation of tRNA and rRNA 5-methylcytosine in the kingdom Plantae.BMC Plant Biol. 2015 Aug 14;15:199. doi: 10.1186/s12870-015-0580-8. BMC Plant Biol. 2015. PMID: 26268215 Free PMC article.
-
Pseudouridylation meets next-generation sequencing.Methods. 2016 Sep 1;107:63-72. doi: 10.1016/j.ymeth.2016.03.001. Epub 2016 Mar 8. Methods. 2016. PMID: 26968262 Review.
-
Analysis of RNA Modifications by Second- and Third-Generation Deep Sequencing: 2020 Update.Genes (Basel). 2021 Feb 16;12(2):278. doi: 10.3390/genes12020278. Genes (Basel). 2021. PMID: 33669207 Free PMC article. Review.
Cited by
-
New Twists in Detecting mRNA Modification Dynamics.Trends Biotechnol. 2021 Jan;39(1):72-89. doi: 10.1016/j.tibtech.2020.06.002. Epub 2020 Jul 1. Trends Biotechnol. 2021. PMID: 32620324 Free PMC article. Review.
-
Detection of ribonucleoside modifications by liquid chromatography coupled with mass spectrometry.Biochim Biophys Acta Gene Regul Mech. 2019 Mar;1862(3):280-290. doi: 10.1016/j.bbagrm.2018.10.012. Epub 2018 Nov 7. Biochim Biophys Acta Gene Regul Mech. 2019. PMID: 30414470 Free PMC article. Review.
-
5-Methylcytosine profiles in mouse transcriptomes suggest the randomness of m5C formation catalyzed by RNA methyltransferase.BMC Res Notes. 2022 Feb 23;15(1):81. doi: 10.1186/s13104-022-05968-7. BMC Res Notes. 2022. PMID: 35197120 Free PMC article.
-
Dnmt2 mediates intergenerational transmission of paternally acquired metabolic disorders through sperm small non-coding RNAs.Nat Cell Biol. 2018 May;20(5):535-540. doi: 10.1038/s41556-018-0087-2. Epub 2018 Apr 25. Nat Cell Biol. 2018. PMID: 29695786 Free PMC article.
-
Distinguishing RNA modifications from noise in epitranscriptome maps.Nat Chem Biol. 2018 Feb 14;14(3):215-225. doi: 10.1038/nchembio.2546. Nat Chem Biol. 2018. PMID: 29443978
References
-
- Adams JM, Cory S. 1975. Modified nucleosides and bizarre 5′-termini in mouse myeloma mRNA. Nature 255: 28–33. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases