

Endothelial Cell Autophagy Maintains Shear Stress-Induced Nitric Oxide Generation via Glycolysis-Dependent Purinergic Signaling to Endothelial Nitric Oxide Synthase
- PMID: 28684613
- PMCID: PMC5693355
- DOI: 10.1161/ATVBAHA.117.309510
Endothelial Cell Autophagy Maintains Shear Stress-Induced Nitric Oxide Generation via Glycolysis-Dependent Purinergic Signaling to Endothelial Nitric Oxide Synthase
Abstract
Objective: Impaired endothelial cell (EC) autophagy compromises shear stress-induced nitric oxide (NO) generation. We determined the responsible mechanism.
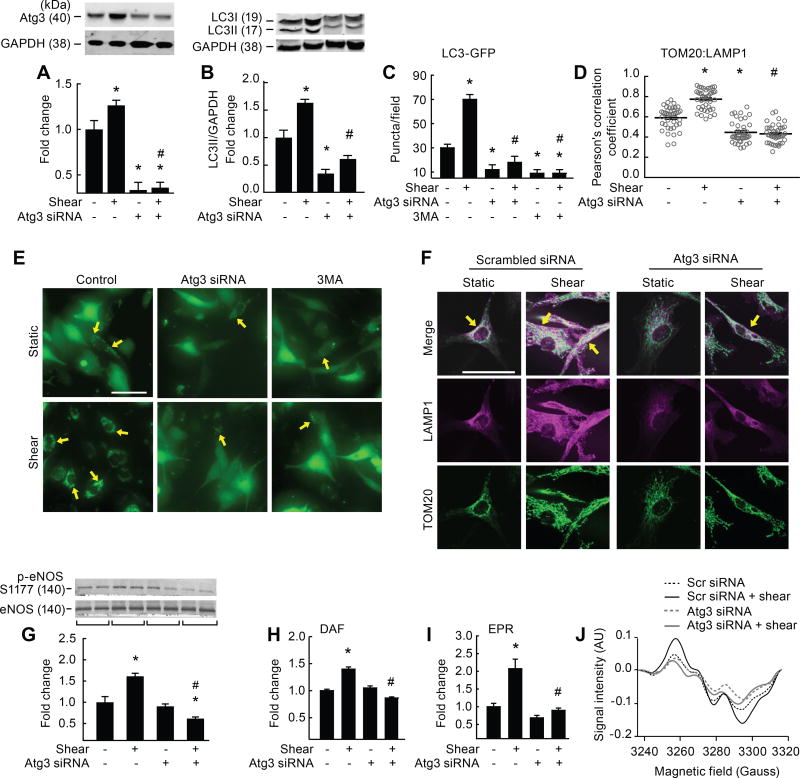
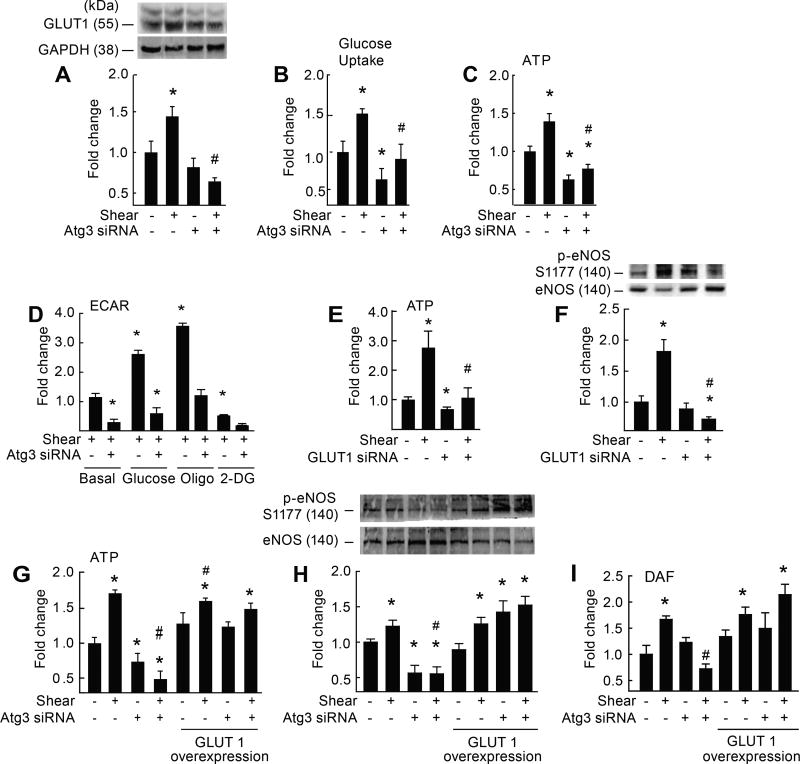
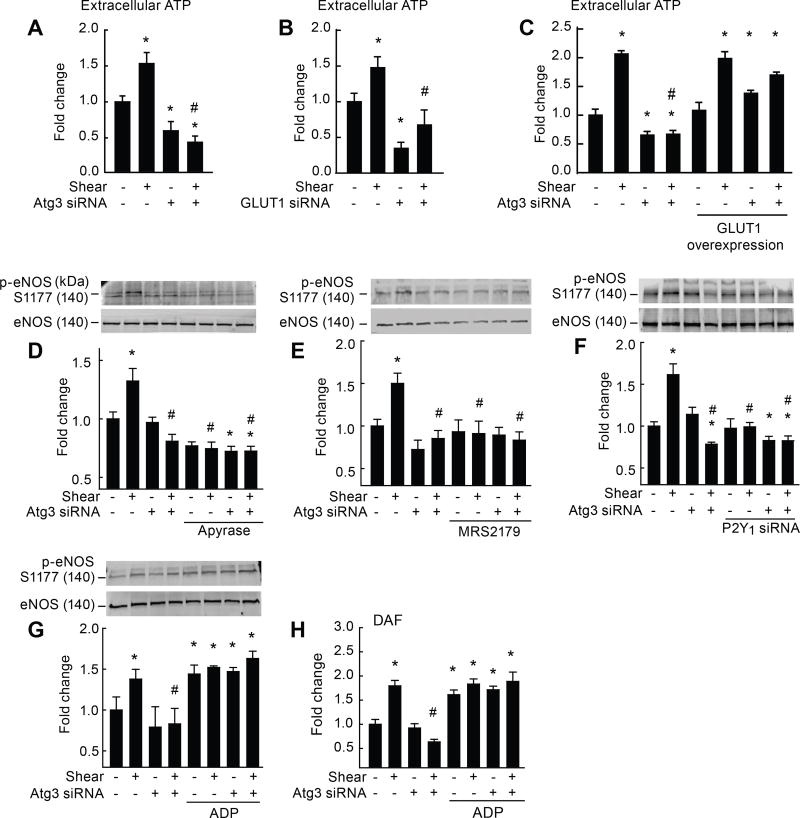
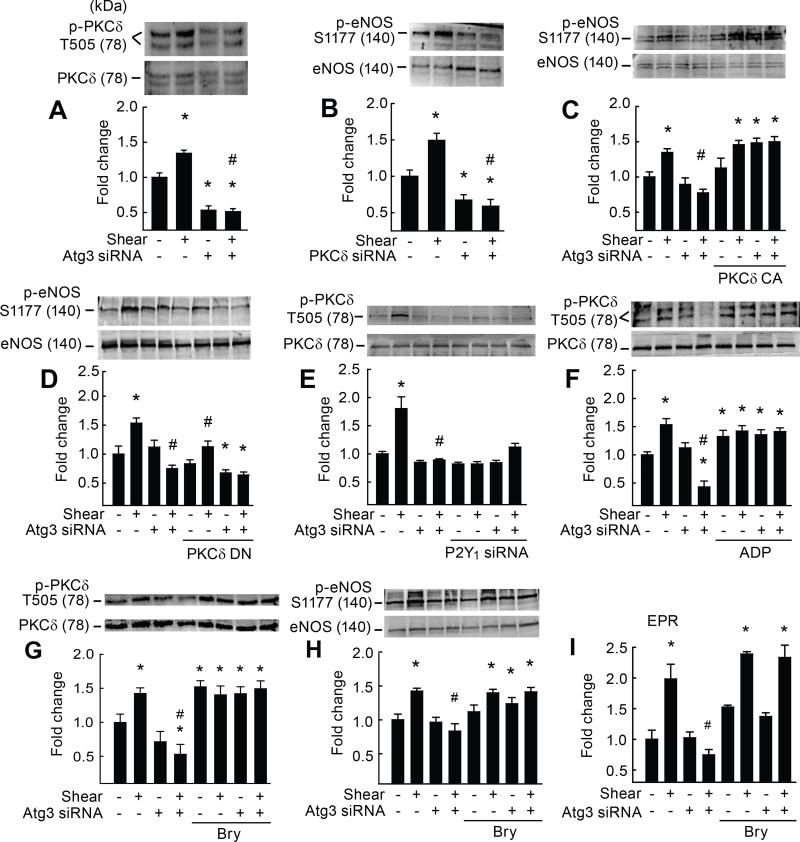
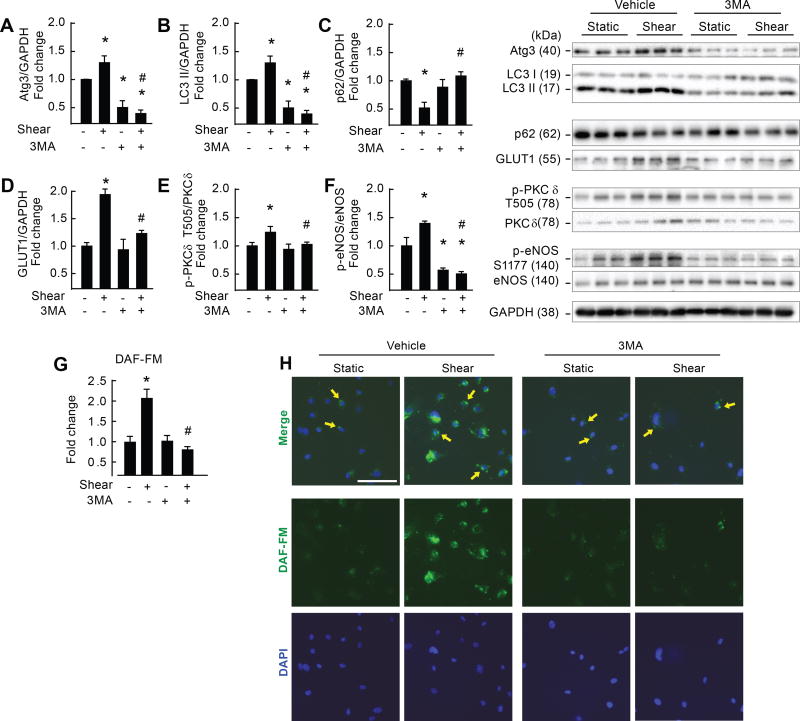
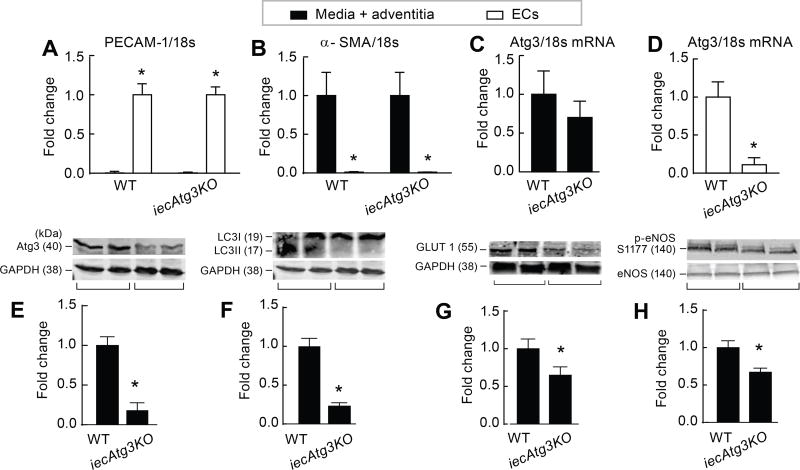
Approach and results: On autophagy compromise in bovine aortic ECs exposed to shear stress, a decrease in glucose uptake and EC glycolysis attenuated ATP production. We hypothesized that decreased glycolysis-dependent purinergic signaling via P2Y1 (P2Y purinoceptor 1) receptors, secondary to impaired autophagy in ECs, prevents shear-induced phosphorylation of eNOS (endothelial nitric oxide synthase) at its positive regulatory site S1117 (p-eNOSS1177) and NO generation. Maneuvers that restore glucose transport and glycolysis (eg, overexpression of GLUT1 [glucose transporter 1]) or purinergic signaling (eg, addition of exogenous ADP) rescue shear-induced p-eNOSS1177 and NO production in ECs with impaired autophagy. Conversely, inhibiting glucose transport via GLUT1 small interfering RNA, blocking purinergic signaling via ectonucleotidase-mediated ATP/ADP degradation (eg, apyrase), or inhibiting P2Y1 receptors using pharmacological (eg, MRS2179 [2'-deoxy-N6-methyladenosine 3',5'-bisphosphate tetrasodium salt]) or genetic (eg, P2Y1-receptor small interfering RNA) procedures inhibit shear-induced p-eNOSS1177 and NO generation in ECs with intact autophagy. Supporting a central role for PKCδT505 (protein kinase C delta T505) in relaying the autophagy-dependent purinergic-mediated signal to eNOS, we find that (1) shear stress-induced activating phosphorylation of PKCδT505 is negated by inhibiting autophagy, (2) shear-induced p-eNOSS1177 and NO generation are restored in autophagy-impaired ECs via pharmacological (eg, bryostatin) or genetic (eg, constitutively active PKCδ) activation of PKCδT505, and (3) pharmacological (eg, rottlerin) and genetic (eg, PKCδ small interfering RNA) PKCδ inhibition prevents shear-induced p-eNOSS1177 and NO generation in ECs with intact autophagy. Key nodes of dysregulation in this pathway on autophagy compromise were revealed in human arterial ECs.
Conclusions: Targeted reactivation of purinergic signaling and PKCδ has strategic potential to restore compromised NO generation in pathologies associated with suppressed EC autophagy.
Keywords: autophagy; cell physiological phenomena; endothelial cells; nitric oxide; reactive oxygen species.
© 2017 American Heart Association, Inc.
Figures
References
-
- Zhang Y, Janssens SP, Wingler K, Schmidt HH, Moens AL. Modulating endothelial nitric oxide synthase: a new cardiovascular therapeutic strategy. Am J Physiol Heart Circ. Physiol. 2011;301:H634–H646. - PubMed
-
- Triggle CR, Hollenberg M, Anderson TJ, Ding H, Jiang Y, Ceroni L, Wiehler WB, Ng ESM, Ellis A, Andrews K, McGuire JJ, Pannirselvam M. The endothelium in health and disease- a target for therapeutic intervention. J Sm Musc. 2003;39:249–267. - PubMed
-
- Kuma A, Mizushima N. Physiological role of autophagy as an intracellular recycling system: with an emphasis on nutrient metabolism. Semin Cell Dev Biol. 2010;21:683–690. - PubMed
-
- Mizushima N. Physiological functions of autophagy. Curr Top Microbiol Immunol. 2009;335:71–84. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
