

GPR37L1 modulates seizure susceptibility: Evidence from mouse studies and analyses of a human GPR37L1 variant
- PMID: 28688853
- PMCID: PMC5569905
- DOI: 10.1016/j.nbd.2017.07.006
GPR37L1 modulates seizure susceptibility: Evidence from mouse studies and analyses of a human GPR37L1 variant
Abstract
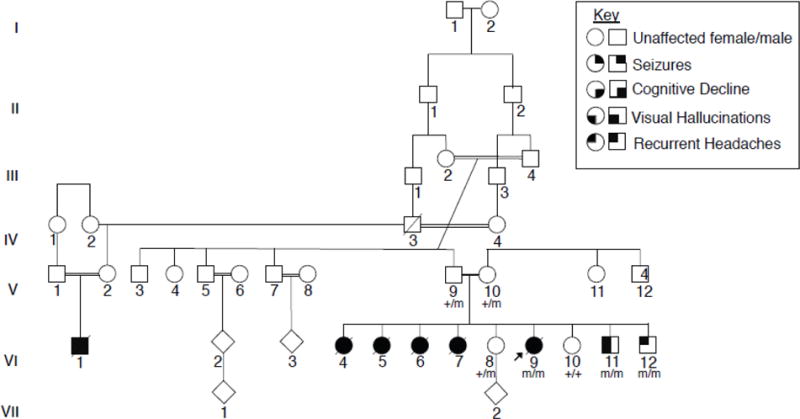
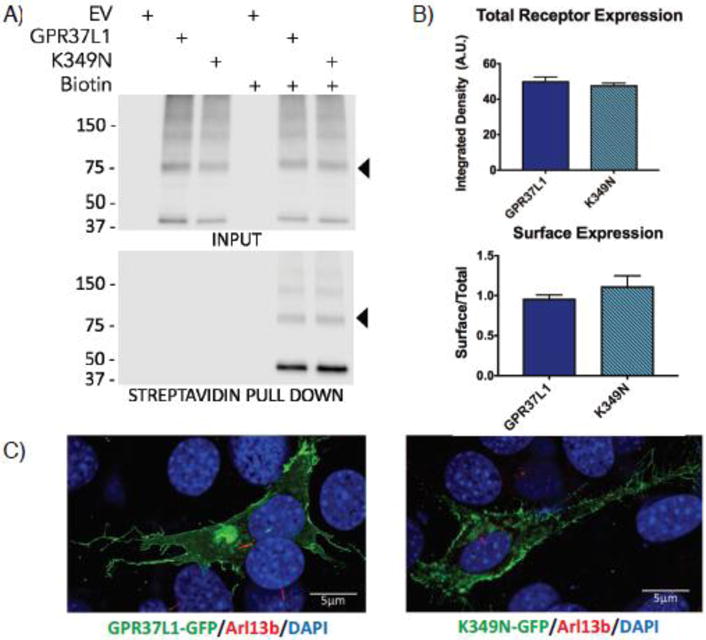
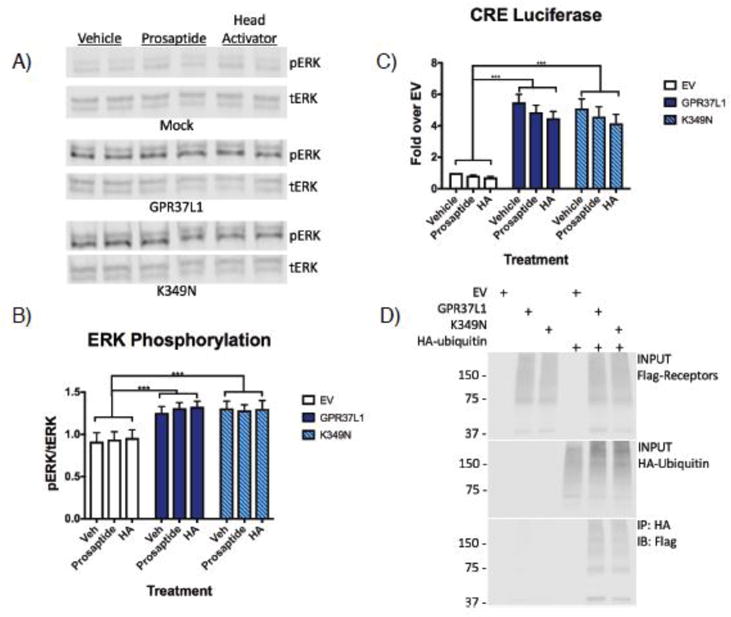
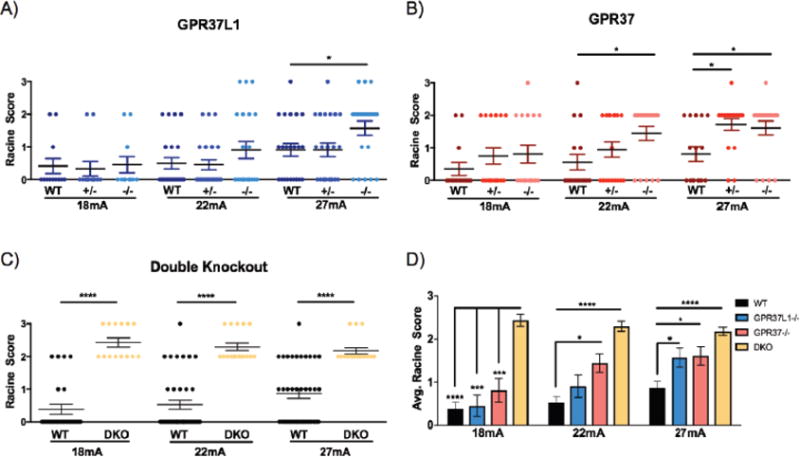
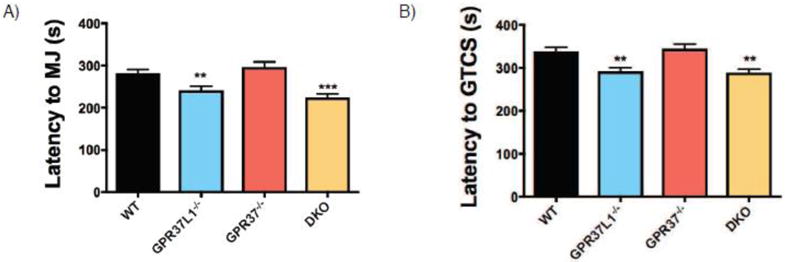
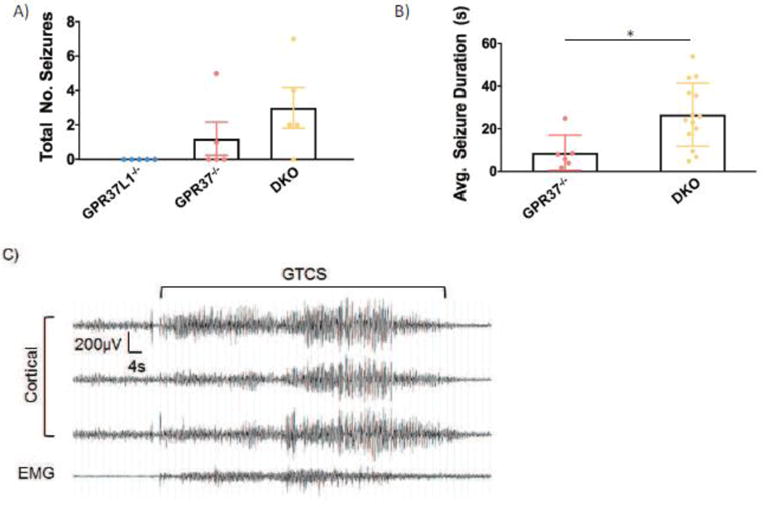
Progressive myoclonus epilepsies (PMEs) are disorders characterized by myoclonic and generalized seizures with progressive neurological deterioration. While several genetic causes for PMEs have been identified, the underlying causes remain unknown for a substantial portion of cases. Here we describe several affected individuals from a large, consanguineous family presenting with a novel PME in which symptoms begin in adolescence and result in death by early adulthood. Whole exome analyses revealed that affected individuals have a homozygous variant in GPR37L1 (c.1047G>T [Lys349Asn]), an orphan G protein-coupled receptor (GPCR) expressed predominantly in the brain. In vitro studies demonstrated that the K349N substitution in Gpr37L1 did not grossly alter receptor expression, surface trafficking or constitutive signaling in transfected cells. However, in vivo studies revealed that a complete loss of Gpr37L1 function in mice results in increased seizure susceptibility. Mice lacking the related receptor Gpr37 also exhibited an increase in seizure susceptibility, while genetic deletion of both receptors resulted in an even more dramatic increase in vulnerability to seizures. These findings provide evidence linking GPR37L1 and GPR37 to seizure etiology and demonstrate an association between a GPR37L1 variant and a novel progressive myoclonus epilepsy.
Keywords: Brain; Disease; Epilepsy; Mutant; Mutation; Orphan GPCR; Receptors.
Copyright © 2017 Elsevier Inc. All rights reserved.
Figures
References
-
- Berg AT, Berkovic SF, Brodie MJ, Buchhalter J, Cross JH, van Emde Boas W, Engel J, French J, Glauser TA, Mathern GW, et al. Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005–2009. Epilepsia. 2010;51:676–685. - PubMed
-
- Consortium E, Consortium EM, Steffens M, Leu C, Ruppert AK, Zara F, Striano P, Robbiano A, Capovilla G, Tinuper P, et al. Genome-wide association analysis of genetic generalized epilepsies implicates susceptibility loci at 1q43, 2p16.1, 2q22.3 and 17q21.32. Hum Mol Genet. 2012;21:5359–5372. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
