

Nuclear Acetyl-CoA Production by ACLY Promotes Homologous Recombination
- PMID: 28689661
- PMCID: PMC5580398
- DOI: 10.1016/j.molcel.2017.06.008
Nuclear Acetyl-CoA Production by ACLY Promotes Homologous Recombination
Abstract
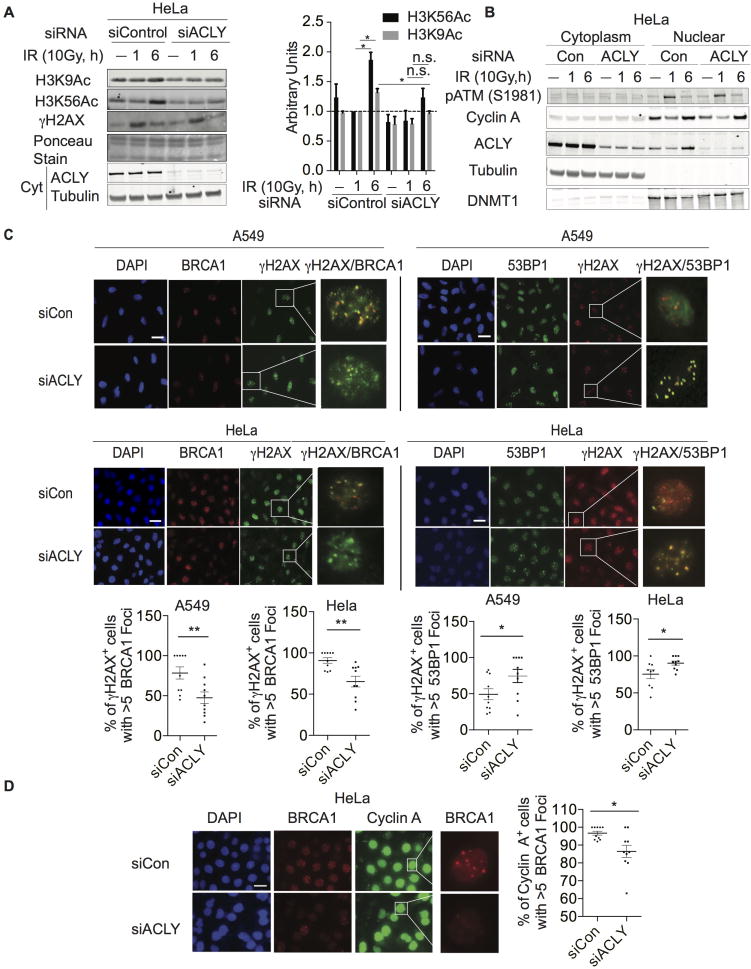
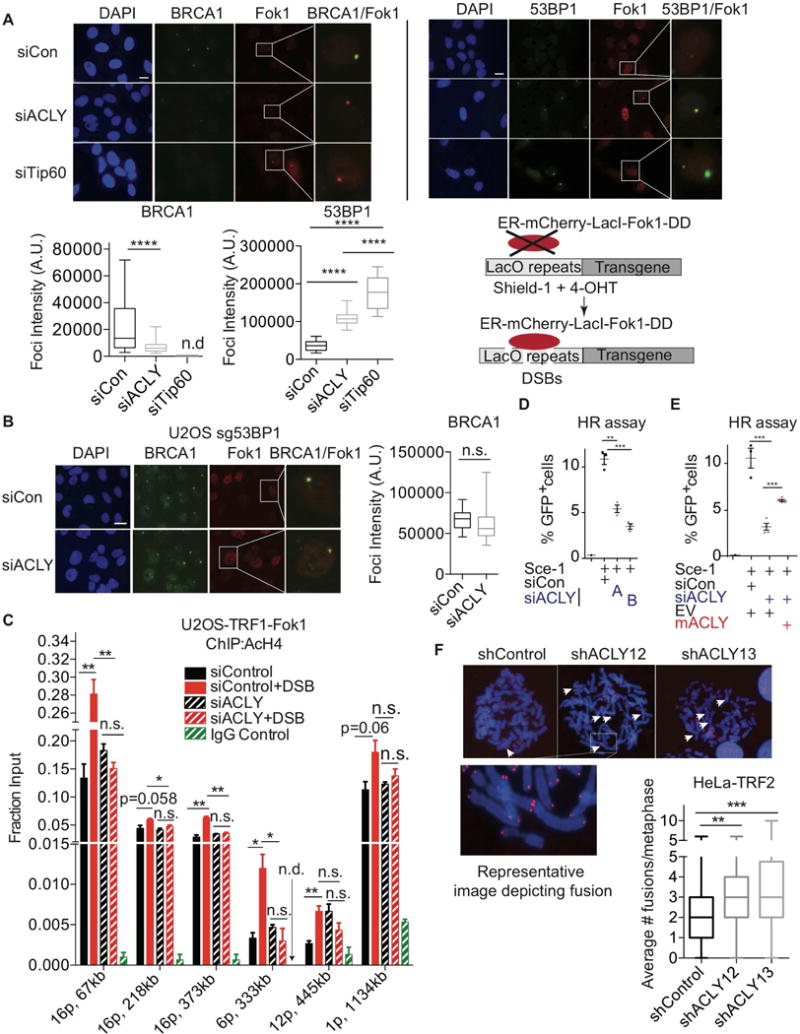
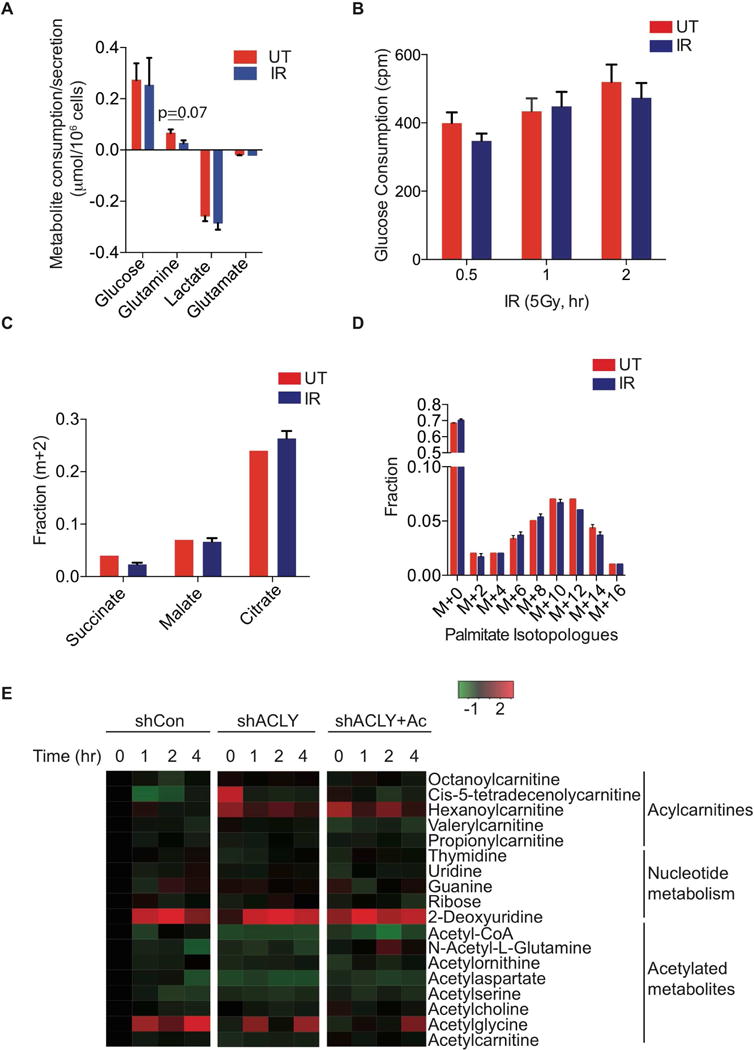
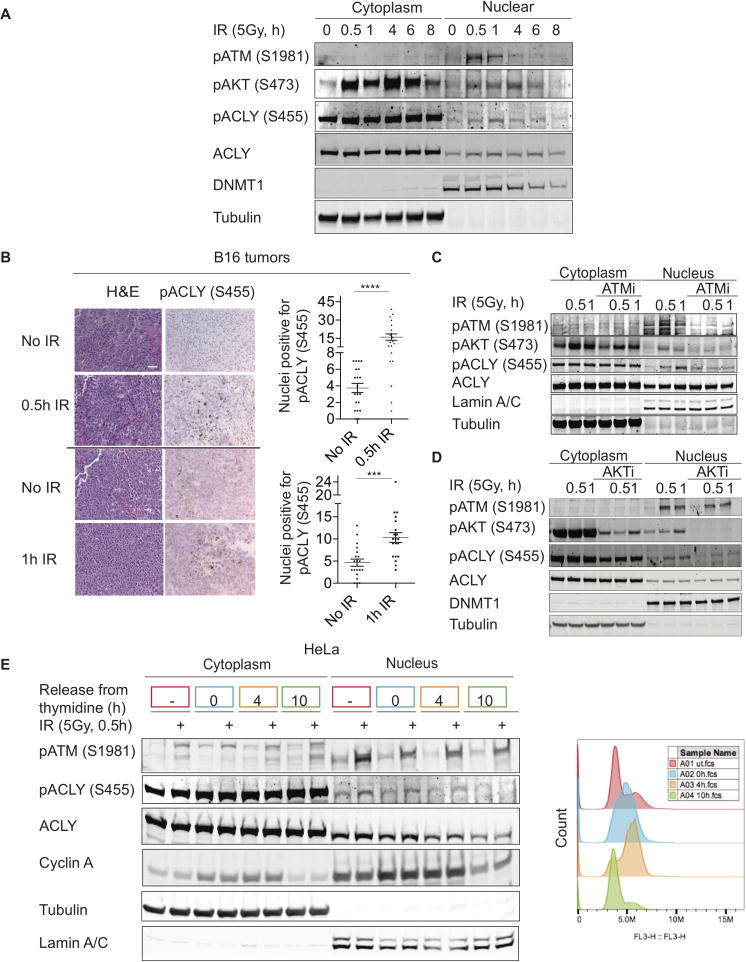
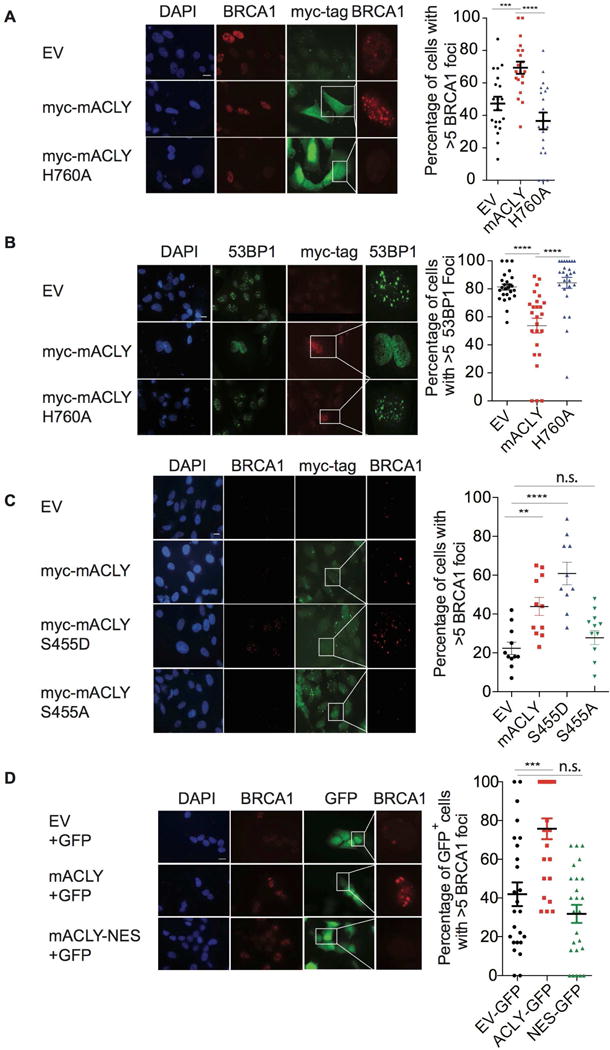
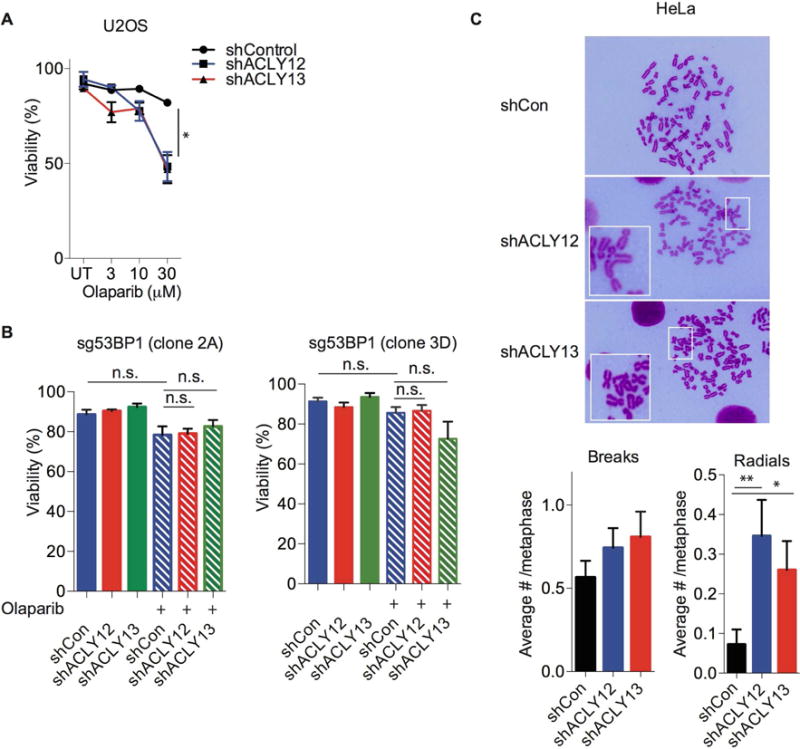
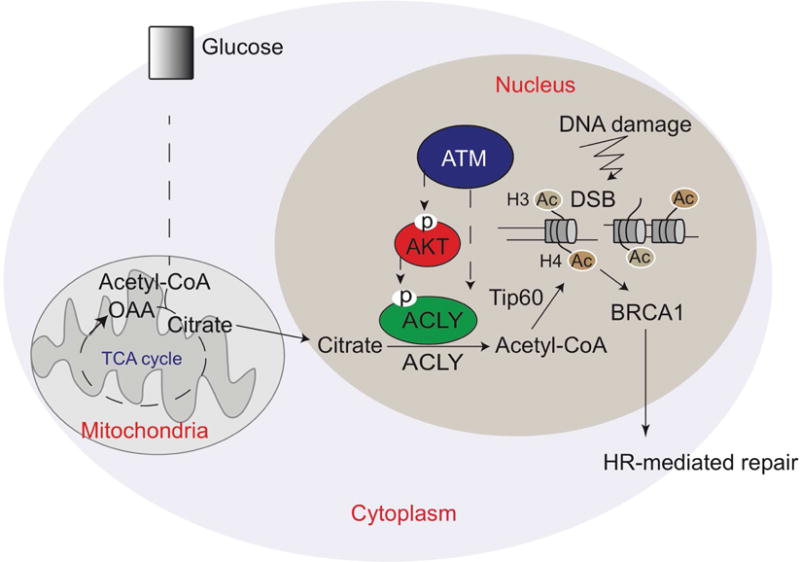
While maintaining the integrity of the genome and sustaining bioenergetics are both fundamental functions of the cell, potential crosstalk between metabolic and DNA repair pathways is poorly understood. Since histone acetylation plays important roles in DNA repair and is sensitive to the availability of acetyl coenzyme A (acetyl-CoA), we investigated a role for metabolic regulation of histone acetylation during the DNA damage response. In this study, we report that nuclear ATP-citrate lyase (ACLY) is phosphorylated at S455 downstream of ataxia telangiectasia mutated (ATM) and AKT following DNA damage. ACLY facilitates histone acetylation at double-strand break (DSB) sites, impairing 53BP1 localization and enabling BRCA1 recruitment and DNA repair by homologous recombination. ACLY phosphorylation and nuclear localization are necessary for its role in promoting BRCA1 recruitment. Upon PARP inhibition, ACLY silencing promotes genomic instability and cell death. Thus, the spatial and temporal control of acetyl-CoA production by ACLY participates in the mechanism of DNA repair pathway choice.
Keywords: ATP-citrate lyase; DNA damage; DNA repair; acetyl-CoA; histone acetylation; homologous recombination; metabolism; non-homologous end joining; nucleus.
Copyright © 2017 Elsevier Inc. All rights reserved.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
-
- Berwick DC, Hers I, Heesom KJ, Moule SK, Tavare JM. The identification of ATP-citrate lyase as a protein kinase B (Akt) substrate in primary adipocytes. J Biol Chem. 2002;277:33895–33900. - PubMed
-
- Bozulic L, Surucu B, Hynx D, Hemmings BA. PKBalpha/Akt1 acts downstream of DNA-PK in the DNA double-strand break response and promotes survival. Mol Cell. 2008;30:203–213. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
