

Bacteriophage evolution differs by host, lifestyle and genome
- PMID: 28692019
- PMCID: PMC5540316
- DOI: 10.1038/nmicrobiol.2017.112
Bacteriophage evolution differs by host, lifestyle and genome
Abstract
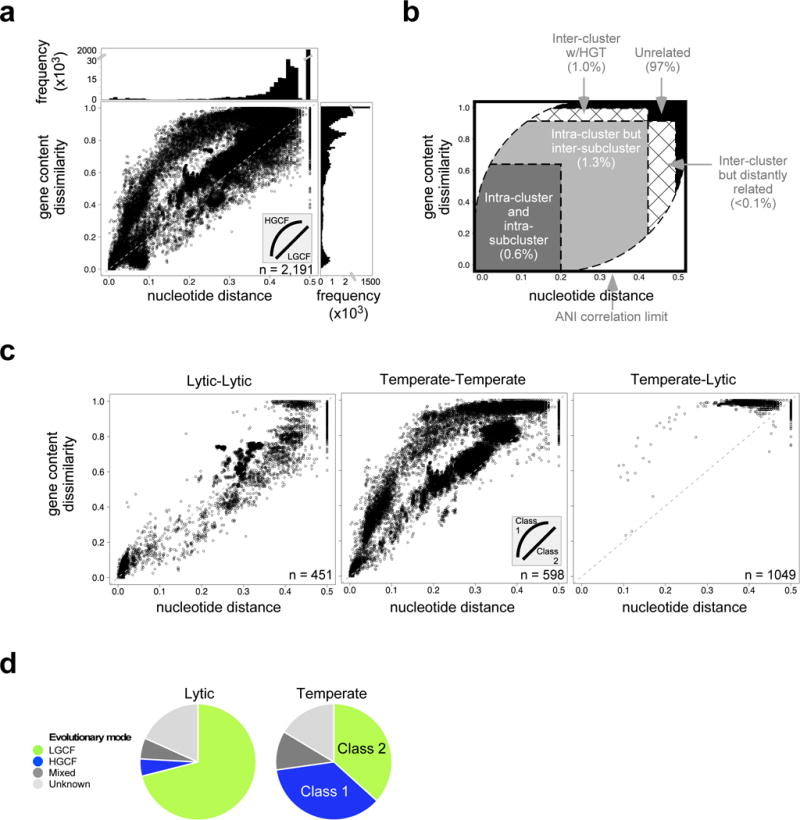
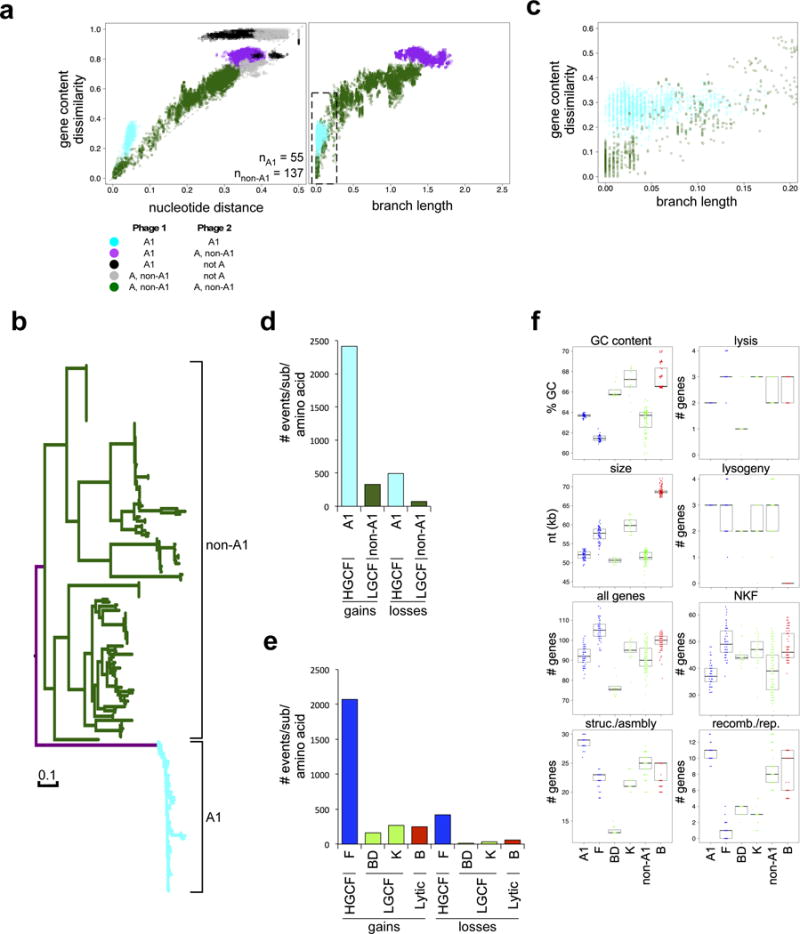
Bacteriophages play key roles in microbial evolution1,2, marine nutrient cycling3 and human disease4. Phages are genetically diverse, and their genome architectures are characteristically mosaic, driven by horizontal gene transfer with other phages and host genomes5. As a consequence, phage evolution is complex and their genomes are composed of genes with distinct and varied evolutionary histories6,7. However, there are conflicting perspectives on the roles of mosaicism and the extent to which it generates a spectrum of genome diversity8 or genetically discrete populations9,10. Here, we show that bacteriophages evolve within two general evolutionary modes that differ in the extent of horizontal gene transfer by an order of magnitude. Temperate phages distribute into high and low gene flux modes, whereas lytic phages share only the lower gene flux mode. The evolutionary modes are also a function of the bacterial host and different proportions of temperate and lytic phages are distributed in either mode depending on the host phylum. Groups of genetically related phages fall into either the high or low gene flux modes, suggesting there are genetic as well as ecological drivers of horizontal gene transfer rates. Consequently, genome mosaicism varies depending on the host, lifestyle and genetic constitution of phages.
Conflict of interest statement
The authors declare no competing financial interests.
Figures


References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
