

Review
doi: 10.1039/c4sc01945h.
Epub 2015 Feb 12.
Molecular engineering of mechanophore activity for stress-responsive polymeric materials
Affiliations
- PMID: 28694949
- PMCID: PMC5485571
- DOI: 10.1039/c4sc01945h
Item in Clipboard
Review
Molecular engineering of mechanophore activity for stress-responsive polymeric materials
Chem Sci.
.
Abstract
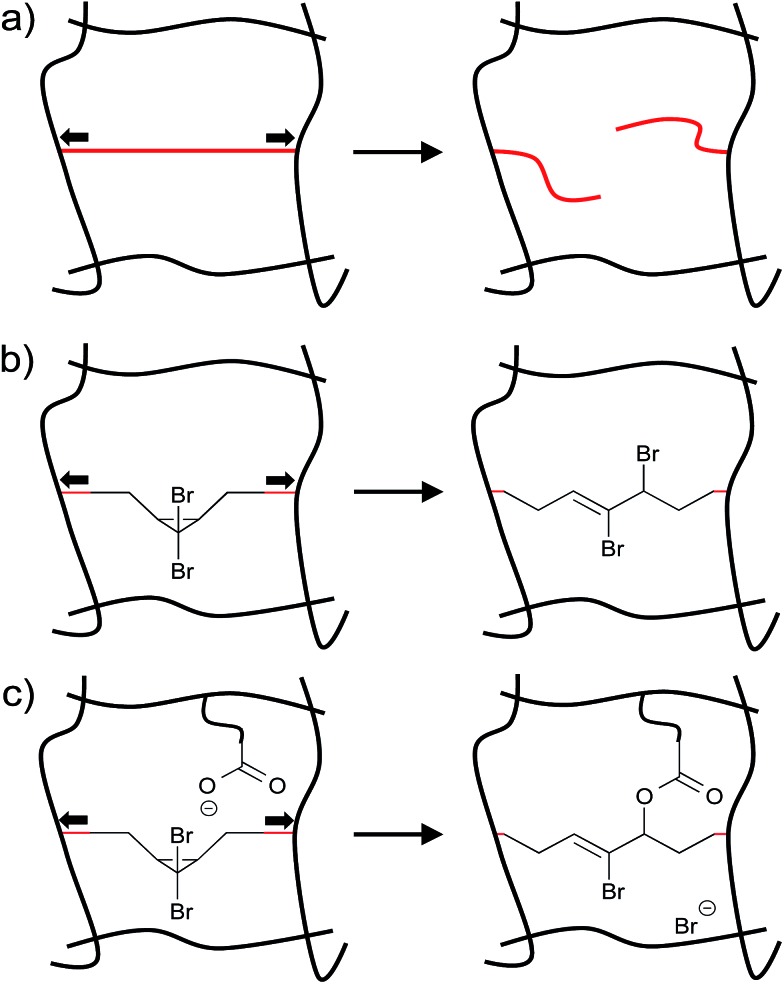
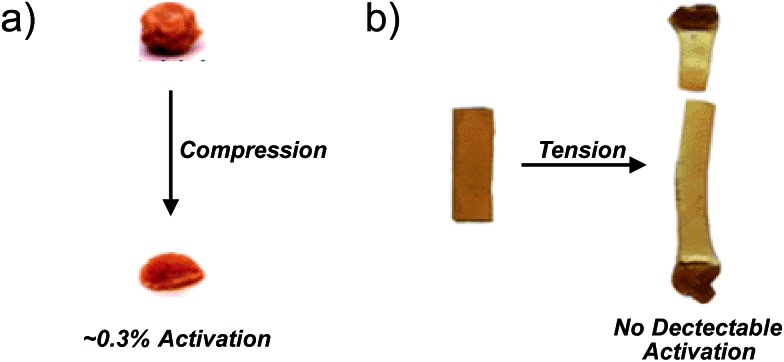
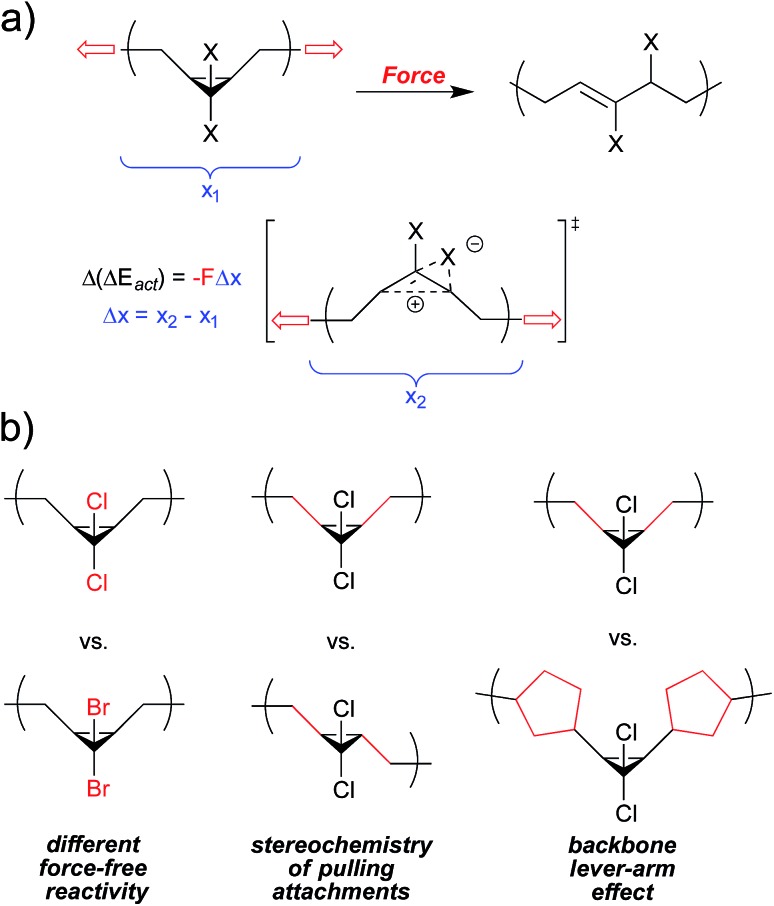
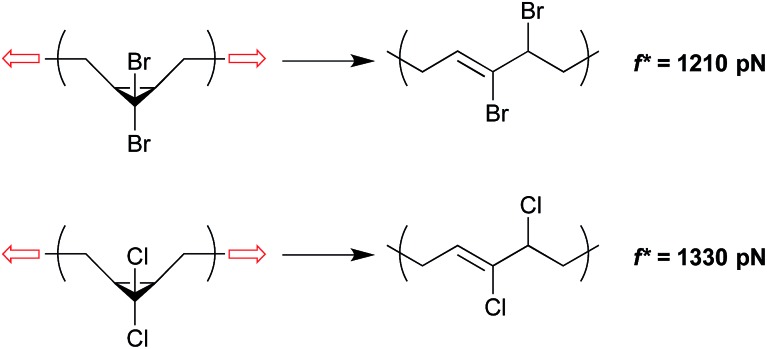
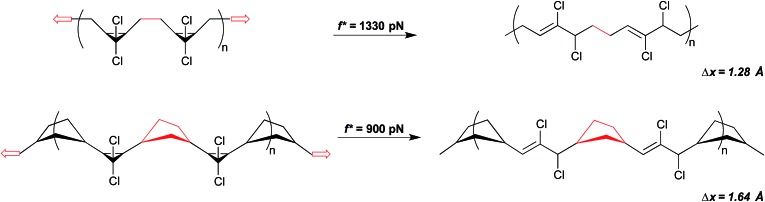
Force reactive functional groups, or mechanophores, have emerged as the basis of a potential strategy for sensing and countering stress-induced material failure. The general utility of this strategy is limited, however, because the levels of mechanophore activation in the bulk are typically low and observed only under large, typically irreversible strains. Strategies that enhance activation are therefore quite useful. Molecular-level design principles by which to engineer enhanced mechanophore activity are reviewed, with an emphasis on quantitative structure-activity studies determined for a family of gem-dihalocyclopropane mechanophores.
Figures
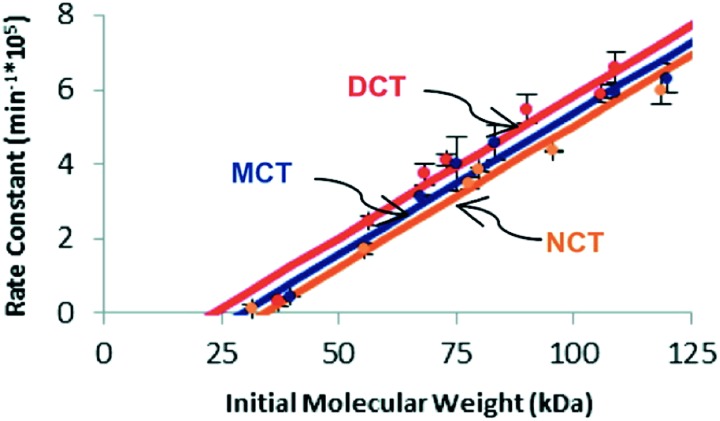
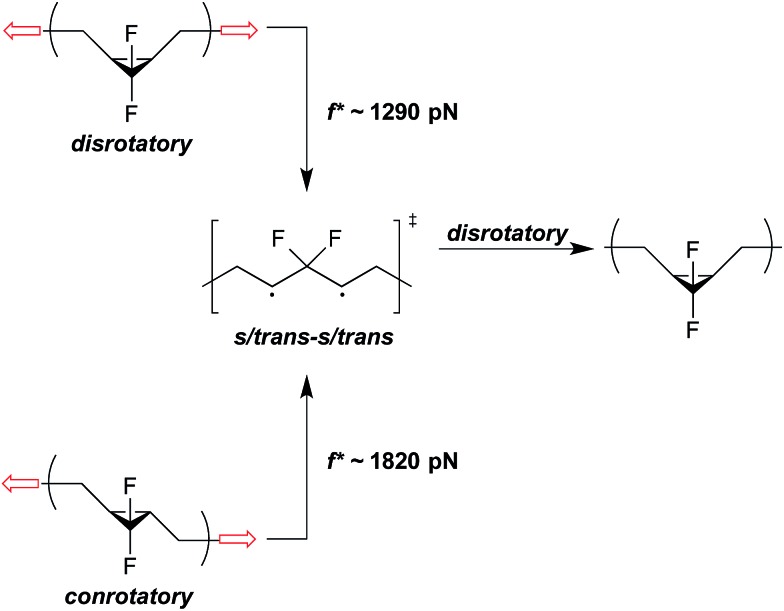
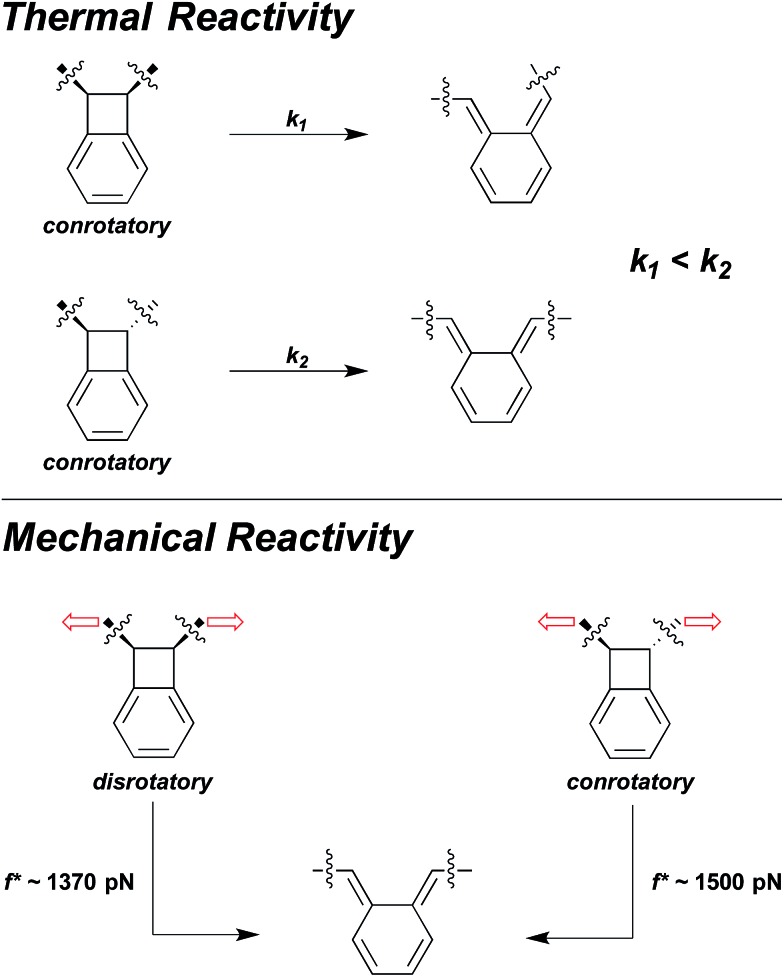
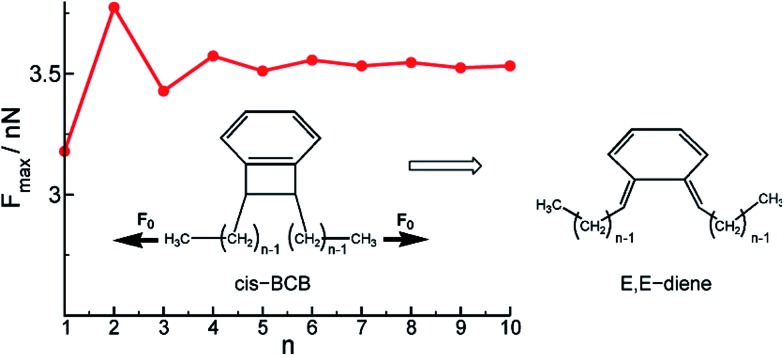

Cameron L. Brown

Stephen L. Craig
References
-
- Beyer M. K., Clausen-Schaumann H. Chem. Rev. 2005;105:2921. - PubMed
-
- Caruso M. M., Davis D. A., Shen Q., Odom S. A., Sottos N. R., White S. R., Moore J. S. Chem. Rev. 2009;109:5755. - PubMed
-
- Black A. L., Lenhardt J. M., Craig S. L. J. Mater. Chem. 2011;21:1655.
-
- Tashiro K., Wu G., Kobayashi M. Polymer. 1988;29:1768.
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials