

Insulin action and resistance in obesity and type 2 diabetes
- PMID: 28697184
- PMCID: PMC6048953
- DOI: 10.1038/nm.4350
Insulin action and resistance in obesity and type 2 diabetes
Abstract
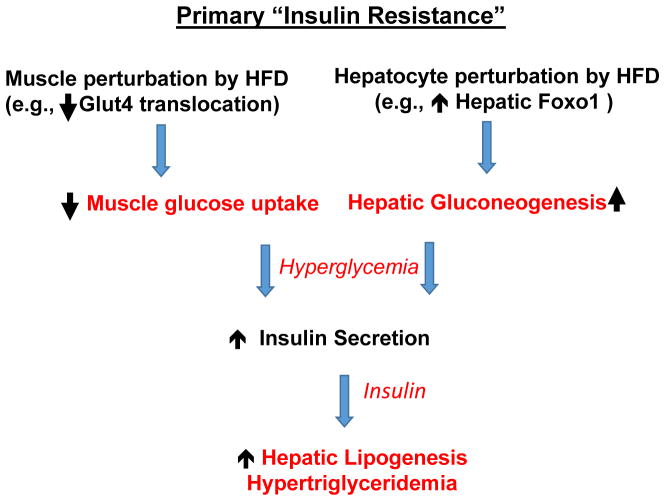
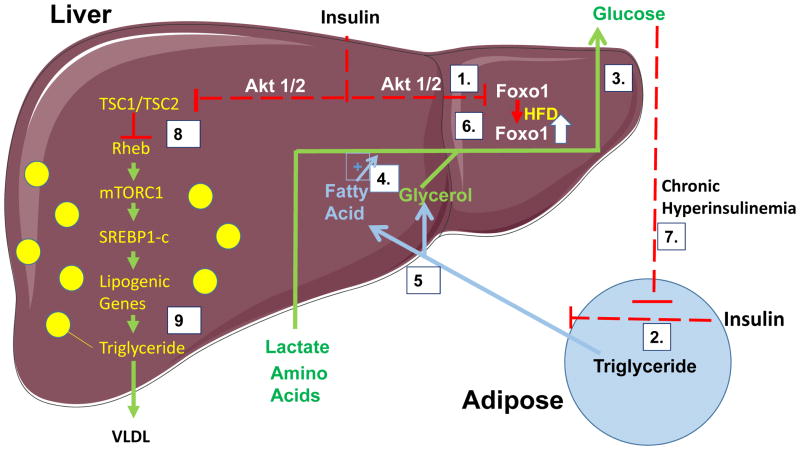
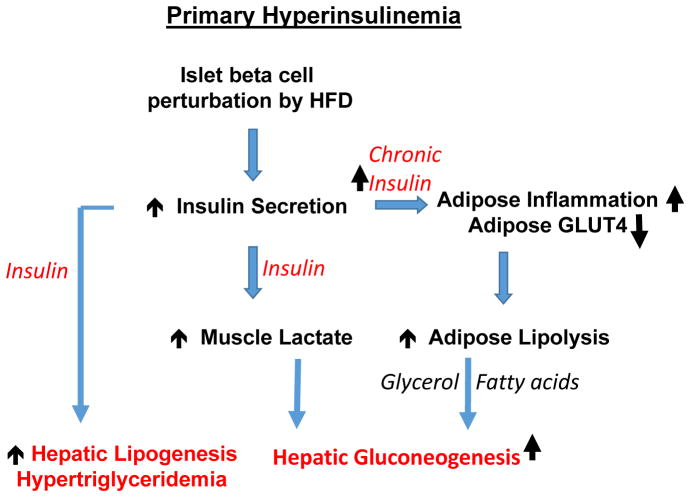
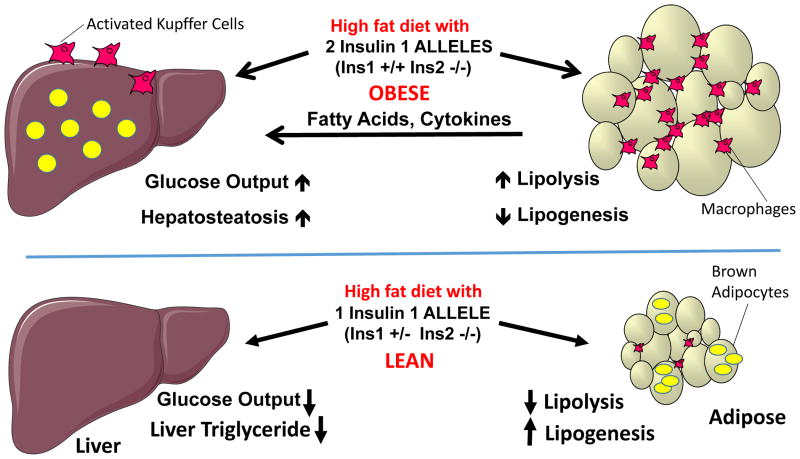
Nutritional excess is a major forerunner of type 2 diabetes. It enhances the secretion of insulin, but attenuates insulin's metabolic actions in the liver, skeletal muscle and adipose tissue. However, conflicting evidence indicates a lack of knowledge of the timing of these events during the development of obesity and diabetes, pointing to a key gap in our understanding of metabolic disease. This Perspective reviews alternate viewpoints and recent results on the temporal and mechanistic connections between hyperinsulinemia, obesity and insulin resistance. Although much attention has addressed early steps in the insulin signaling cascade, insulin resistance in obesity seems to be largely elicited downstream of these steps. New findings also connect insulin resistance to extensive metabolic cross-talk between the liver, adipose tissue, pancreas and skeletal muscle. These and other advances over the past 5 years offer exciting opportunities and daunting challenges for the development of new therapeutic strategies for the treatment of type 2 diabetes.
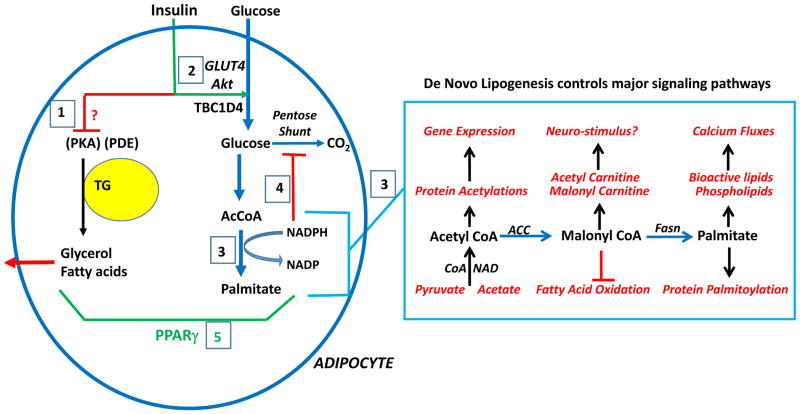
Figures
References
-
- Reaven GM. The insulin resistance syndrome: definition and dietary approaches to treatment. Annu Rev Nutr. 2005;25:391–406. - PubMed
-
- National Diabetes Statistics Report. 2014 www.CDC.gov.
-
- Kloting N, et al. Insulin-sensitive obesity. Am J Physiol Endocrinol Metab. 2010;299:E506–515. - PubMed
-
- DeFronzo RA, Bonadonna RC, Ferrannini E. Pathogenesis of NIDDM. A balanced overview. Diabetes Care. 1992;15:318–368. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
