

Oncogenic Activation of the RNA Binding Protein NELFE and MYC Signaling in Hepatocellular Carcinoma
- PMID: 28697339
- PMCID: PMC5539779
- DOI: 10.1016/j.ccell.2017.06.002
Oncogenic Activation of the RNA Binding Protein NELFE and MYC Signaling in Hepatocellular Carcinoma
Abstract
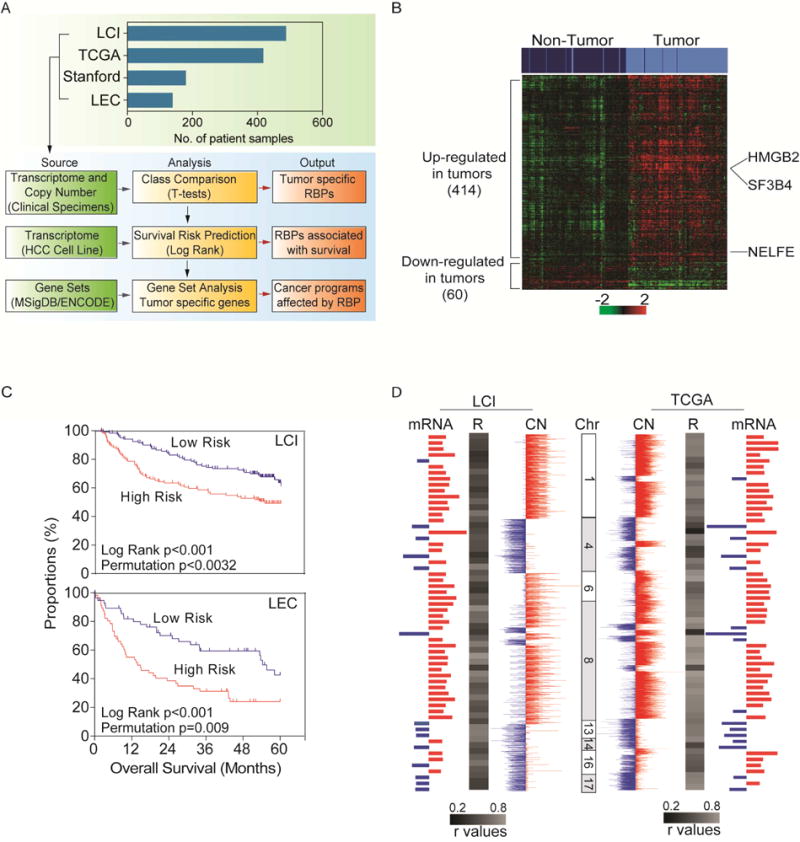
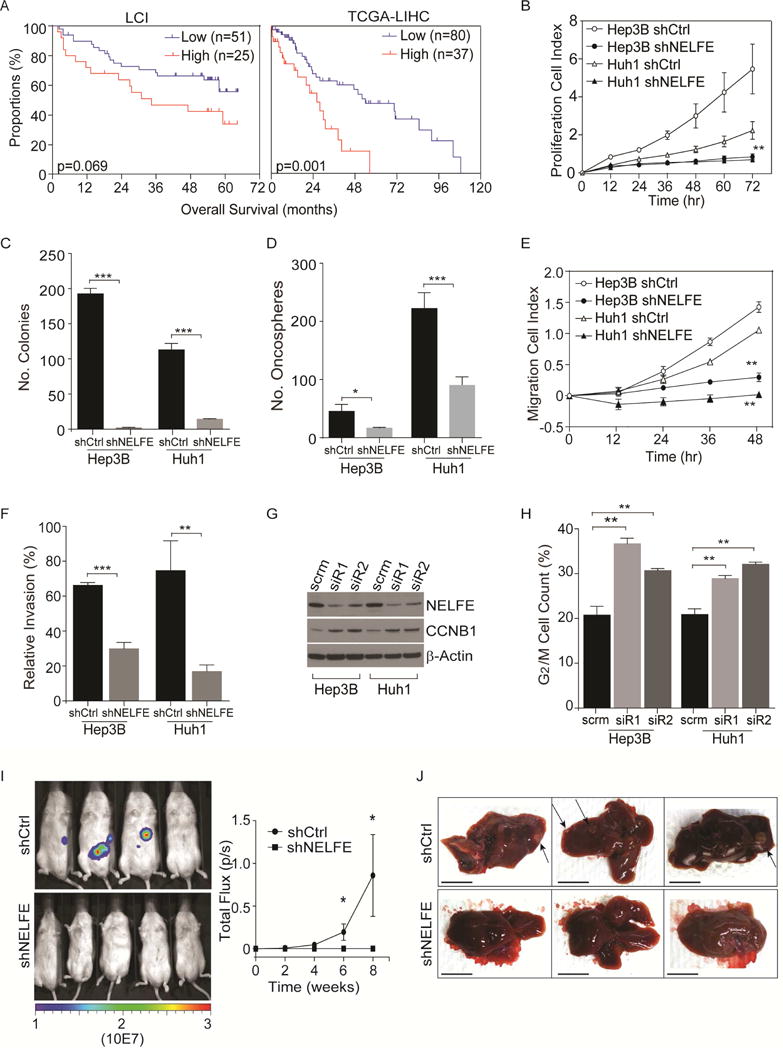
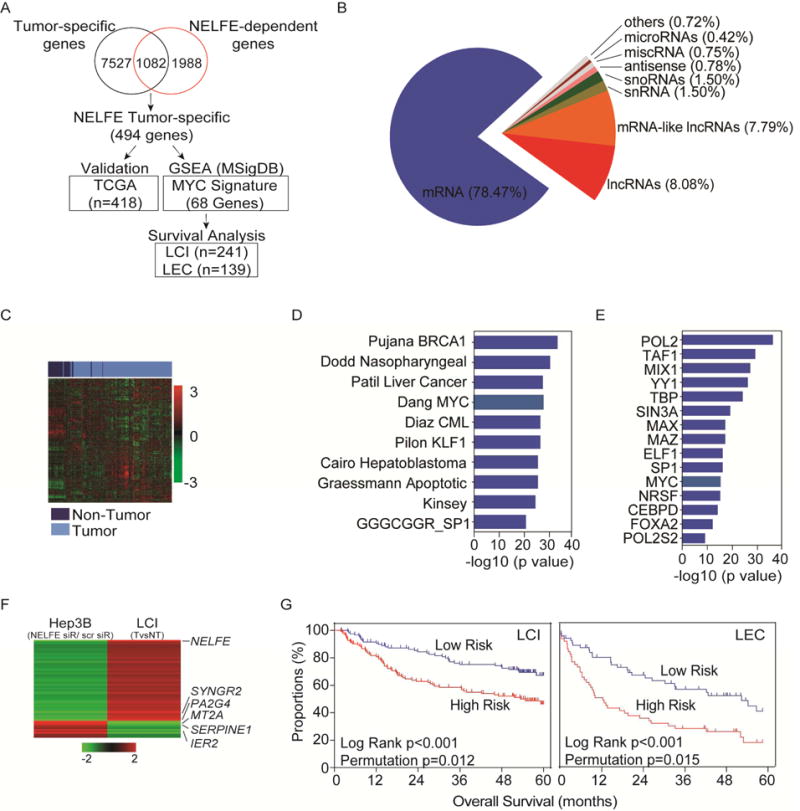
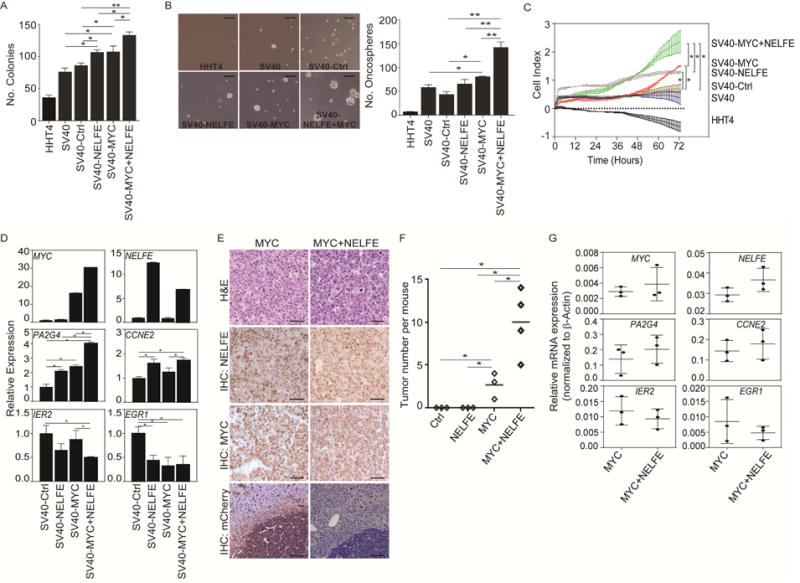
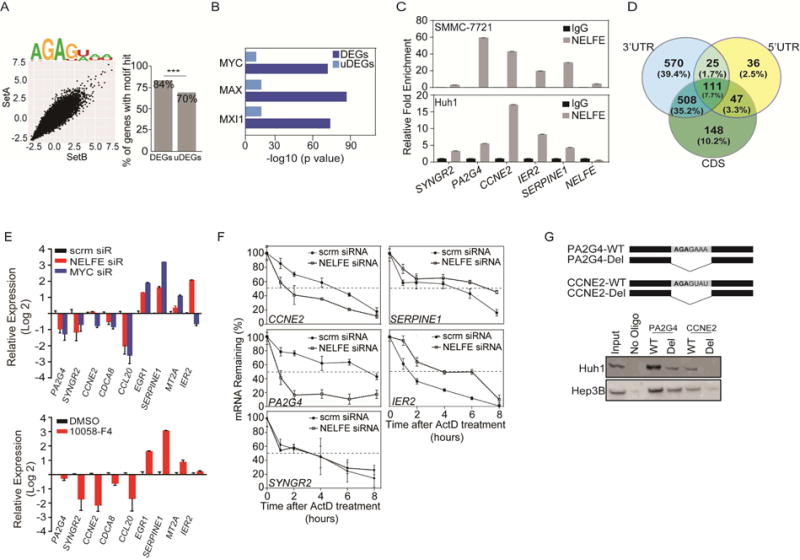
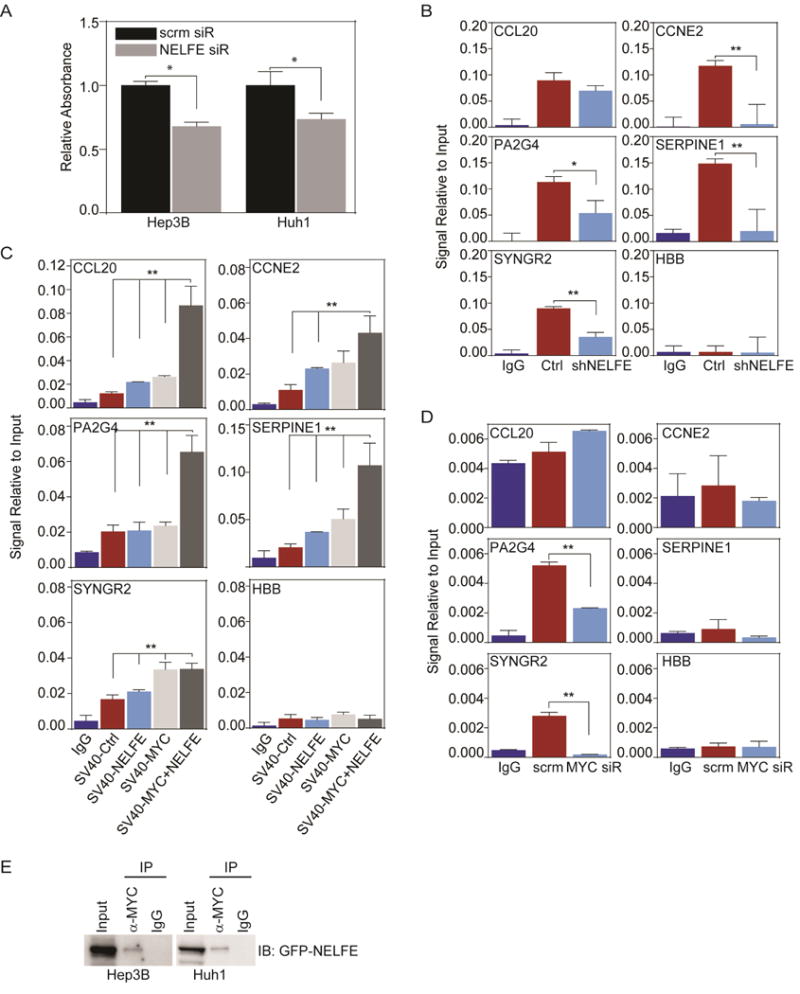
Global transcriptomic imbalance is a ubiquitous feature associated with cancer, including hepatocellular carcinoma (HCC). Analyses of 1,225 clinical HCC samples revealed that a large numbers of RNA binding proteins (RBPs) are dysregulated and that RBP dysregulation is associated with poor prognosis. We further identified that oncogenic activation of a top candidate RBP, negative elongation factor E (NELFE), via somatic copy-number alterations enhanced MYC signaling and promoted HCC progression. Interestingly, NELFE induces a unique tumor transcriptome by selectively regulating MYC-associated genes. Thus, our results revealed NELFE as an oncogenic protein that may contribute to transcriptome imbalance in HCC through the regulation of MYC signaling.
Keywords: MYC; NELFE; RDBP; RNA binding proteins; cancer; hepatocellular carcinoma; oncogene; transcriptome.
Published by Elsevier Inc.
Figures
References
-
- Boyault S, Rickman DS, de Reynies A, Balabaud C, Rebouissou S, Jeannot E, Herault A, Saric J, Belghiti J, Franco D, et al. Transcriptome classification of HCC is related to gene alterations and to new therapeutic targets. Hepatology. 2007;45:42–52. - PubMed
-
- Busa R, Paronetto MP, Farini D, Pierantozzi E, Botti F, Angelini DF, Attisani F, Vespasiani G, Sette C. The RNA-binding protein Sam68 contributes to proliferation and survival of human prostate cancer cells. Oncogene. 2007;26:4372–4382. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
