

MUC1 and HIF-1alpha Signaling Crosstalk Induces Anabolic Glucose Metabolism to Impart Gemcitabine Resistance to Pancreatic Cancer
- PMID: 28697344
- PMCID: PMC5533091
- DOI: 10.1016/j.ccell.2017.06.004
MUC1 and HIF-1alpha Signaling Crosstalk Induces Anabolic Glucose Metabolism to Impart Gemcitabine Resistance to Pancreatic Cancer
Erratum in
-
MUC1 and HIF-1alpha Signaling Crosstalk Induces Anabolic Glucose Metabolism to Impart Gemcitabine Resistance to Pancreatic Cancer.Cancer Cell. 2017 Sep 11;32(3):392. doi: 10.1016/j.ccell.2017.08.008. Cancer Cell. 2017. PMID: 28898700 No abstract available.
Abstract
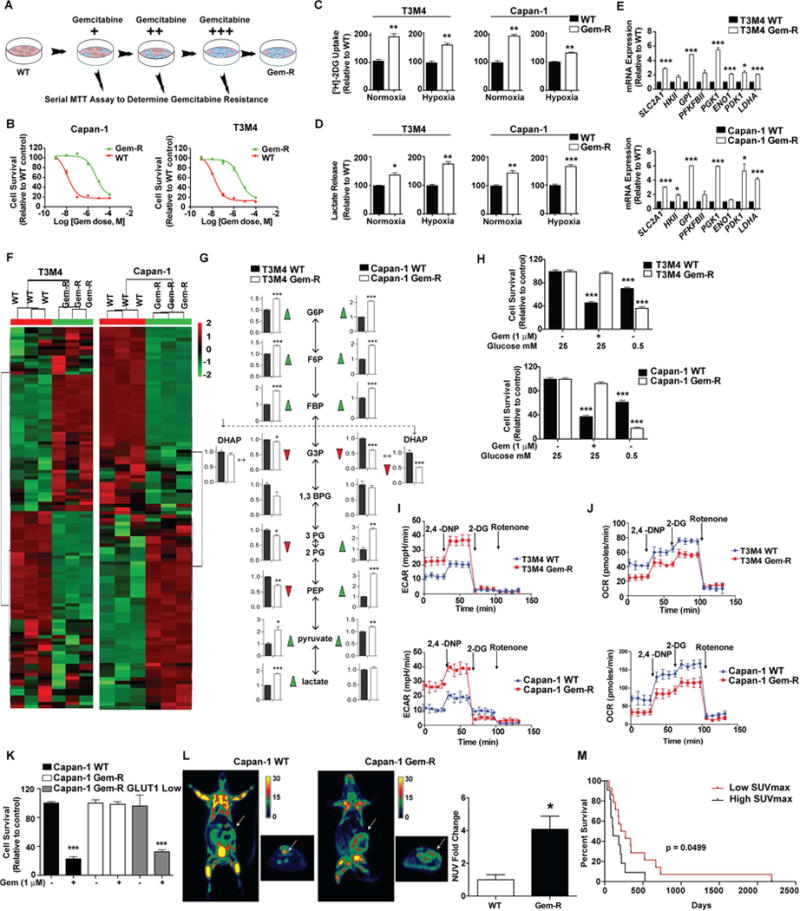
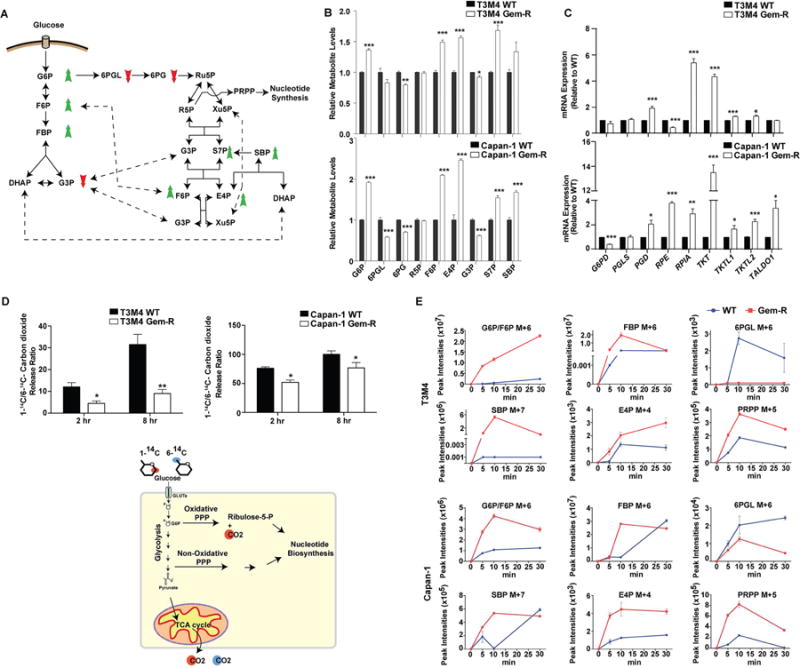
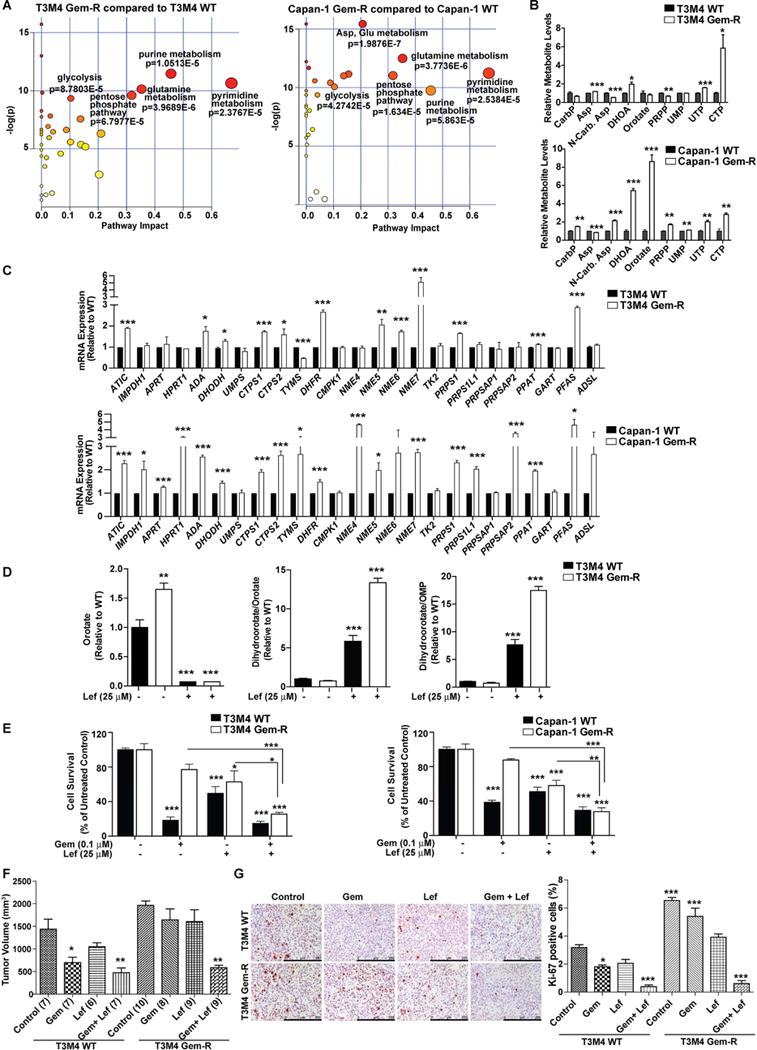
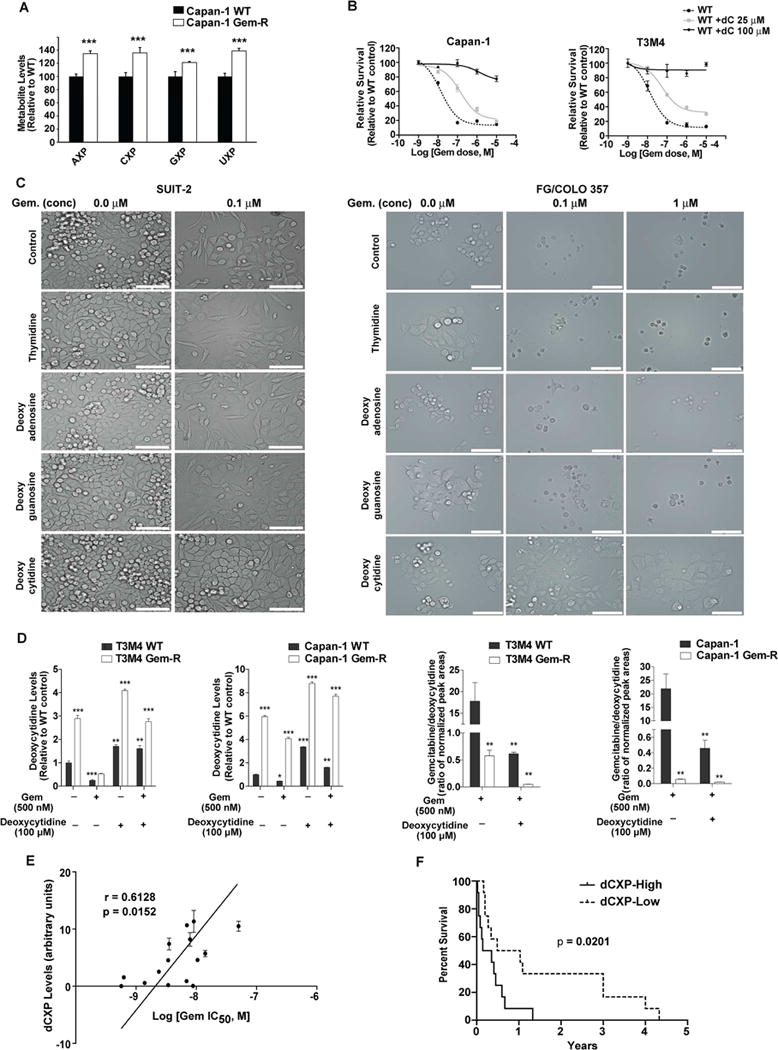
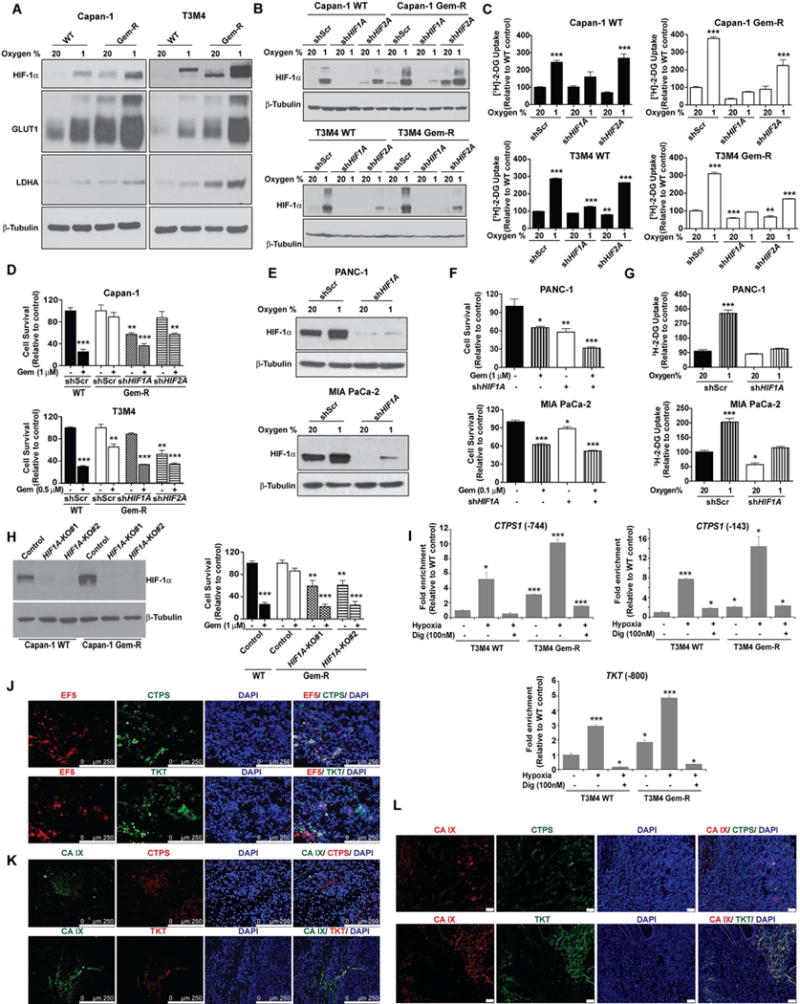
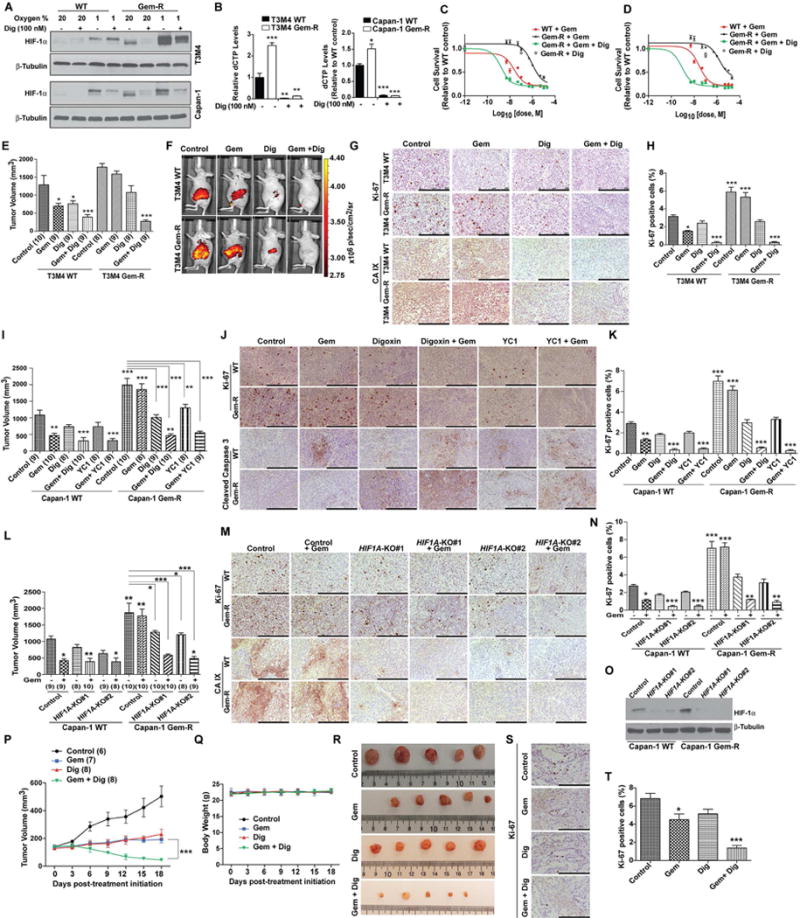
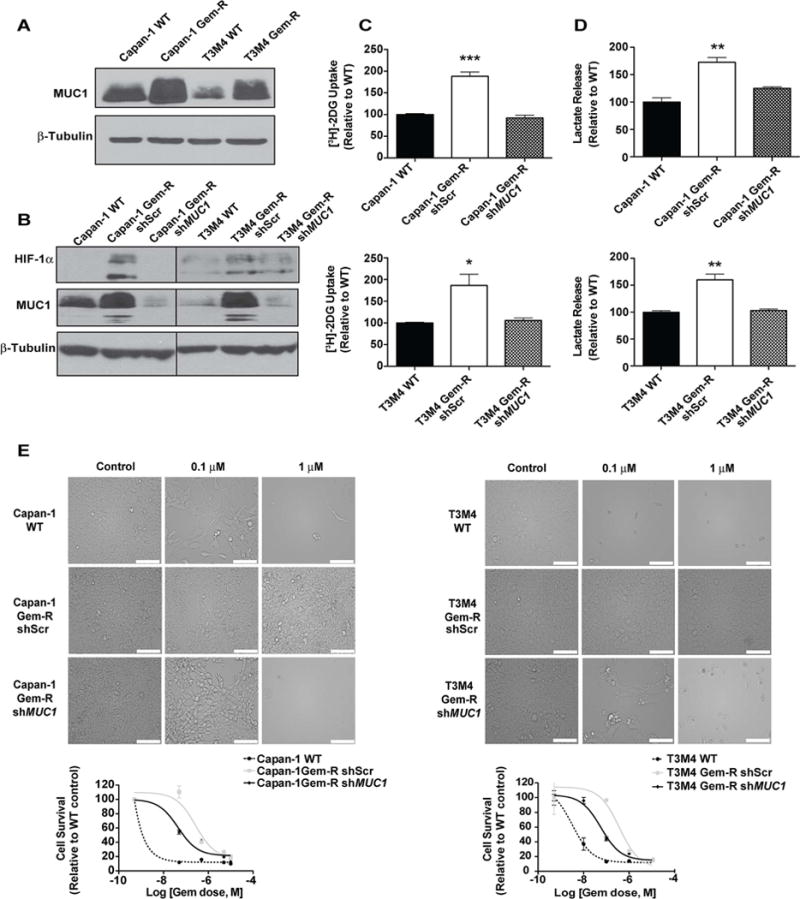
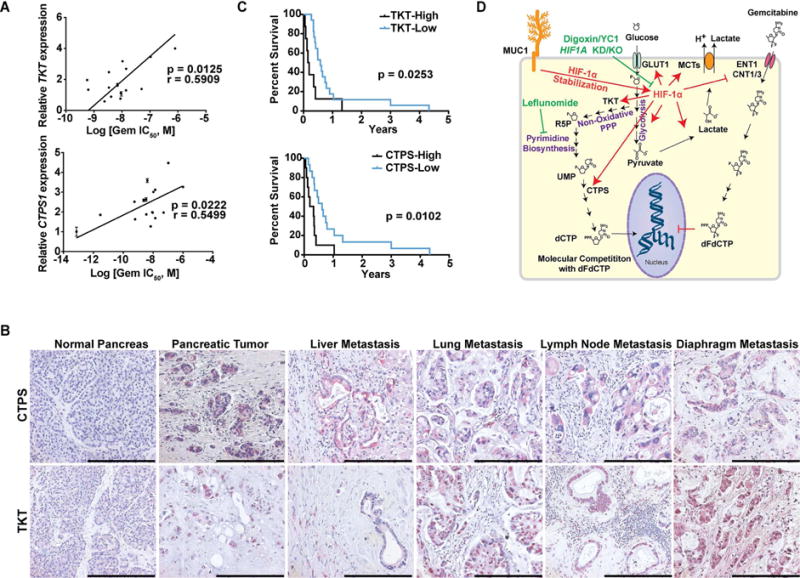
Poor response to cancer therapy due to resistance remains a clinical challenge. The present study establishes a widely prevalent mechanism of resistance to gemcitabine in pancreatic cancer, whereby increased glycolytic flux leads to glucose addiction in cancer cells and a corresponding increase in pyrimidine biosynthesis to enhance the intrinsic levels of deoxycytidine triphosphate (dCTP). Increased levels of dCTP diminish the effective levels of gemcitabine through molecular competition. We also demonstrate that MUC1-regulated stabilization of hypoxia inducible factor-1α (HIF-1α) mediates such metabolic reprogramming. Targeting HIF-1α or de novo pyrimidine biosynthesis, in combination with gemcitabine, strongly diminishes tumor burden. Finally, reduced expression of TKT and CTPS, which regulate flux into pyrimidine biosynthesis, correlates with better prognosis in pancreatic cancer patients on fluoropyrimidine analogs.
Keywords: HIF-1α; MUC1; cancer metabolism; chemotherapy resistance; gemcitabine; mucin; non-oxidative pentose phosphate pathway; nucleotide synthesis; pancreatic cancer; pyrimidine biosynthesis.
Copyright © 2017 Elsevier Inc. All rights reserved.
Conflict of interest statement
Figures
Comment in
-
MUC-king with HIF May Rewire Pyrimidine Biosynthesis and Curb Gemcitabine Resistance in Pancreatic Cancer.Cancer Cell. 2017 Jul 10;32(1):3-5. doi: 10.1016/j.ccell.2017.06.006. Cancer Cell. 2017. PMID: 28697341
References
-
- Akiyama K, Chikayama E, Yuasa H, Shimada Y, Tohge T, Shinozaki K, Hirai MY, Sakurai T, Kikuchi J, Saito K. PRIMe: a Web site that assembles tools for metabolomics and transcriptomics. In silico biology. 2008;8:339–345. - PubMed
-
- Burris H, Storniolo AM. Assessing clinical benefit in the treatment of pancreas cancer: gemcitabine compared to 5-fluorouracil. Eur J Cancer. 1997;33(Suppl 1):S18–22. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
