

Inferring clonal structure in HTLV-1-infected individuals: towards bridging the gap between analysis and visualization
- PMID: 28697807
- PMCID: PMC5505134
- DOI: 10.1186/s40246-017-0112-8
Inferring clonal structure in HTLV-1-infected individuals: towards bridging the gap between analysis and visualization
Abstract
Background: Human T cell leukemia virus type 1 (HTLV-1) causes adult T cell leukemia (ATL) in a proportion of infected individuals after a long latency period. Development of ATL is a multistep clonal process that can be investigated by monitoring the clonal expansion of HTLV-1-infected cells by isolation of provirus integration sites. The clonal composition (size, number, and combinations of clones) during the latency period in a given infected individual has not been clearly elucidated.
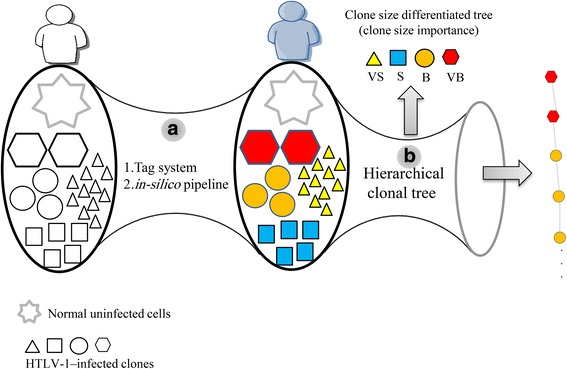
Methods: We used high-throughput sequencing technology coupled with a tag system for isolating integration sites and measuring clone sizes from 60 clinical samples. We assessed the role of clonality and clone size dynamics in ATL onset by modeling data from high-throughput monitoring of HTLV-1 integration sites using single- and multiple-time-point samples.
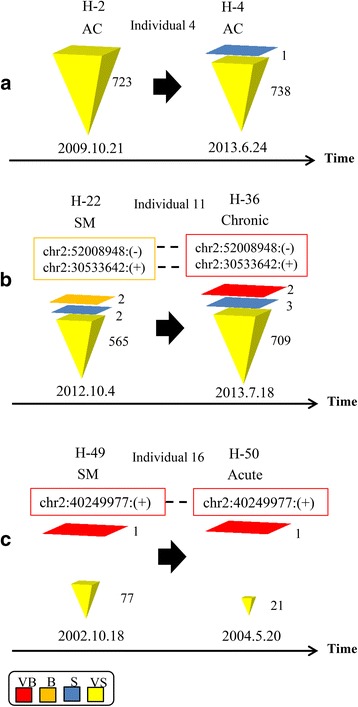
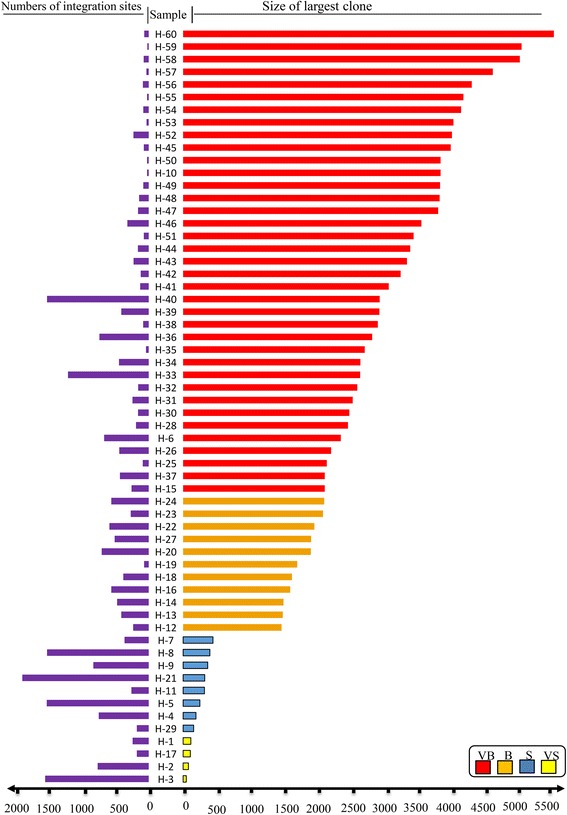
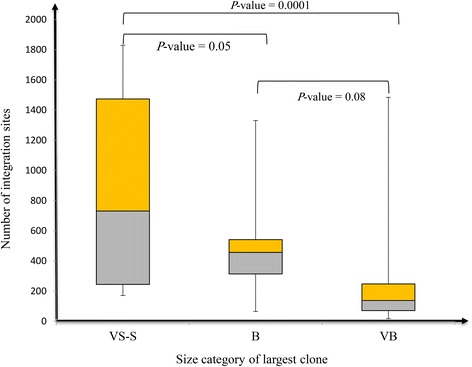
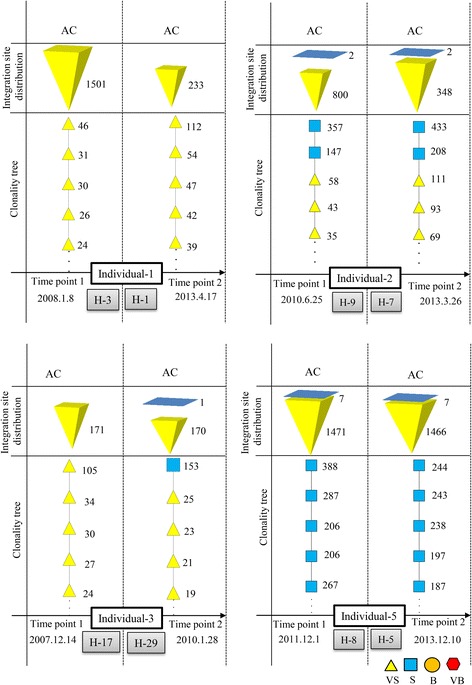
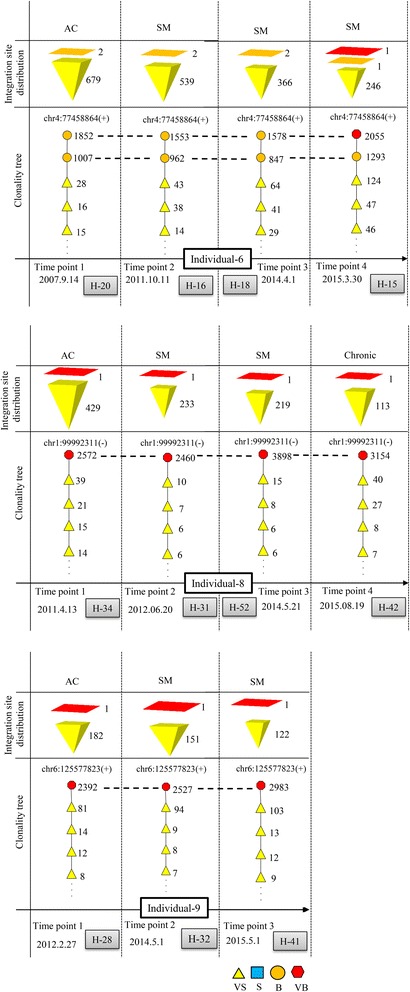
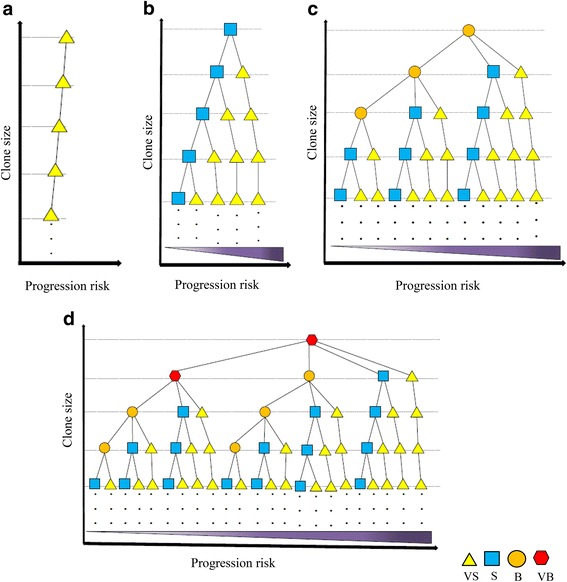
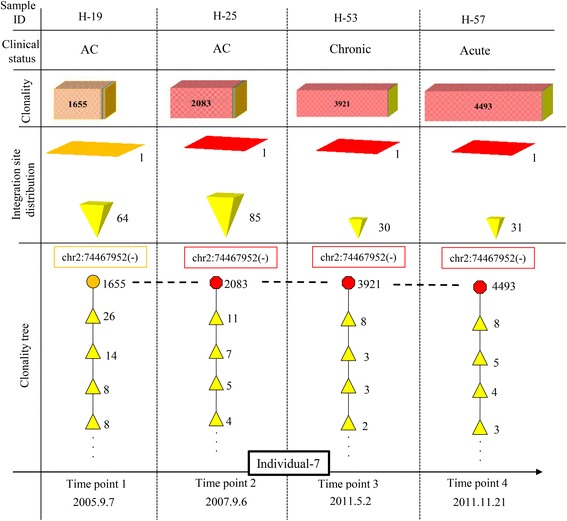
Results: From four size categories analyzed, we found that big clones (B; 513-2048 infected cells) and very big clones (VB; >2048 infected cells) had prognostic value. No sample harbored two or more VB clones or three or more B clones. We examined the role of clone size, clone combination, and the number of integration sites in the prognosis of infected individuals. We found a moderate reverse correlation between the total number of clones and the size of the largest clone. We devised a data-driven model that allows intuitive representation of clonal composition.
Conclusions: This integration site-based clonality tree model represents the complexity of clonality and provides a global view of clonality data that facilitates the analysis, interpretation, understanding, and visualization of the behavior of clones on inter- and intra-individual scales. It is fully data-driven, intuitively depicts the clonality patterns of HTLV-1-infected individuals and can assist in early risk assessment of ATL onset by reflecting the prognosis of infected individuals. This model should assist in assimilating information on clonal composition and understanding clonal expansion in HTLV-1-infected individuals.
Keywords: Adult T cell leukemia; Clonal expansion; Data-driven modeling; High-throughput sequencing; Human T cell leukemia virus type 1; Integration site; Prognostic indicator.
Conflict of interest statement
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
-
- Greaves M. Evolutionary determinants of cancer. Cancer Discov. 2015;5:806–820. doi: 10.1158/2159-8290.CD-15-0439. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
