

Chemistry and Enzymology of Disulfide Cross-Linking in Proteins
- PMID: 28699750
- PMCID: PMC5808910
- DOI: 10.1021/acs.chemrev.7b00123
Chemistry and Enzymology of Disulfide Cross-Linking in Proteins
Abstract
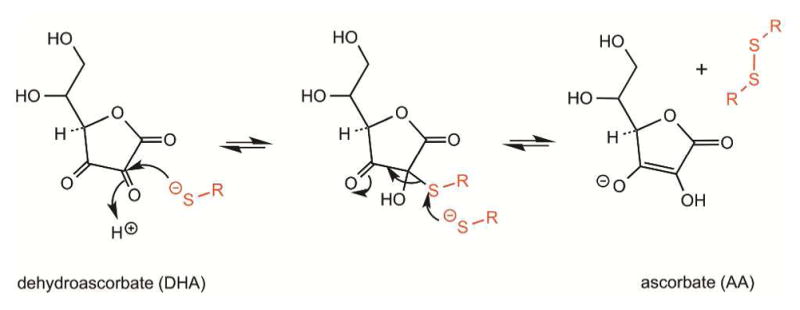
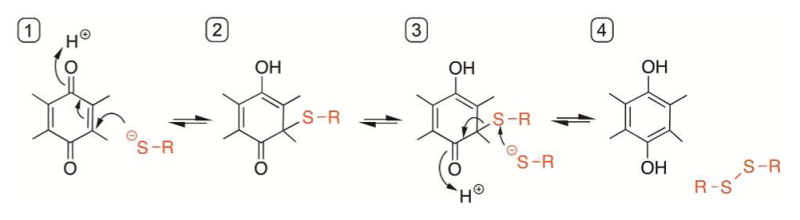
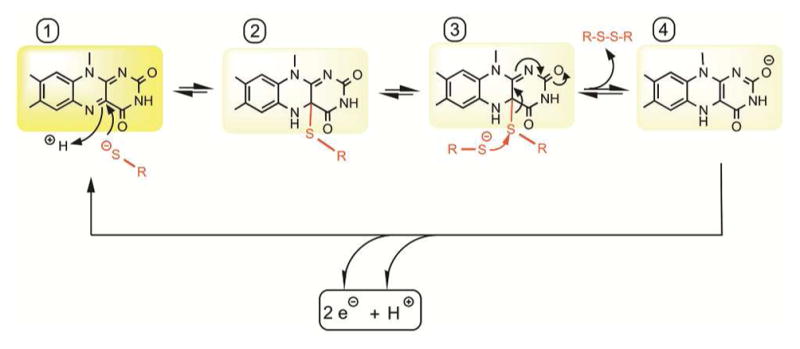
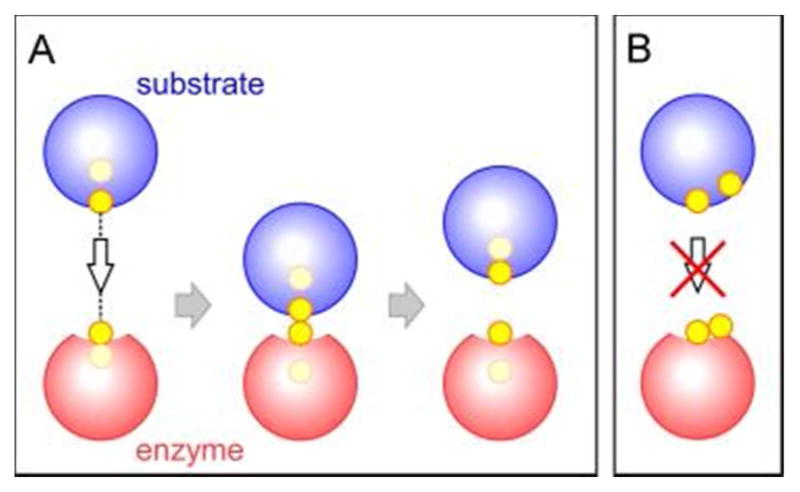
Cysteine thiols are among the most reactive functional groups in proteins, and their pairing in disulfide linkages is a common post-translational modification in proteins entering the secretory pathway. This modest amino acid alteration, the mere removal of a pair of hydrogen atoms from juxtaposed cysteine residues, contrasts with the substantial changes that characterize most other post-translational reactions. However, the wide variety of proteins that contain disulfides, the profound impact of cross-linking on the behavior of the protein polymer, the numerous and diverse players in intracellular pathways for disulfide formation, and the distinct biological settings in which disulfide bond formation can take place belie the simplicity of the process. Here we lay the groundwork for appreciating the mechanisms and consequences of disulfide bond formation in vivo by reviewing chemical principles underlying cysteine pairing and oxidation. We then show how enzymes tune redox-active cofactors and recruit oxidants to improve the specificity and efficiency of disulfide formation. Finally, we discuss disulfide bond formation in a cellular context and identify important principles that contribute to productive thiol oxidation in complex, crowded, dynamic environments.
Figures



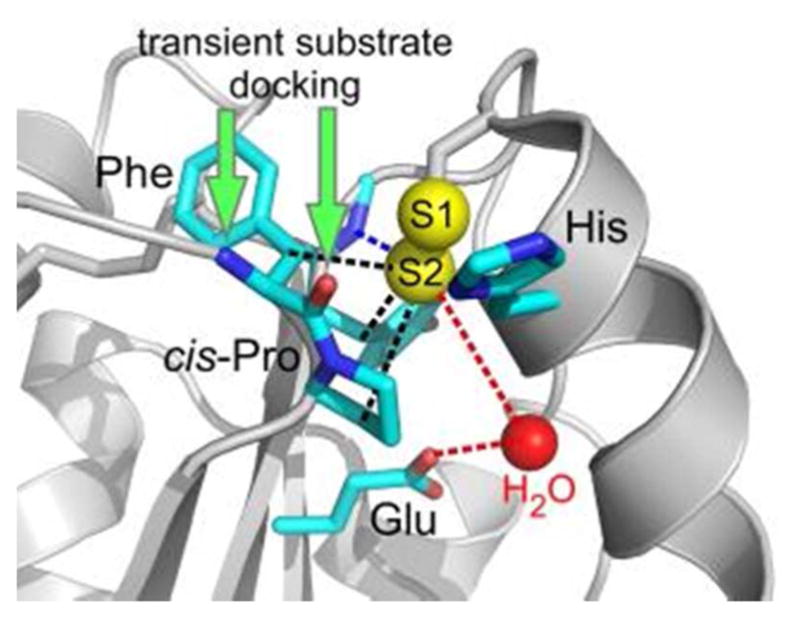
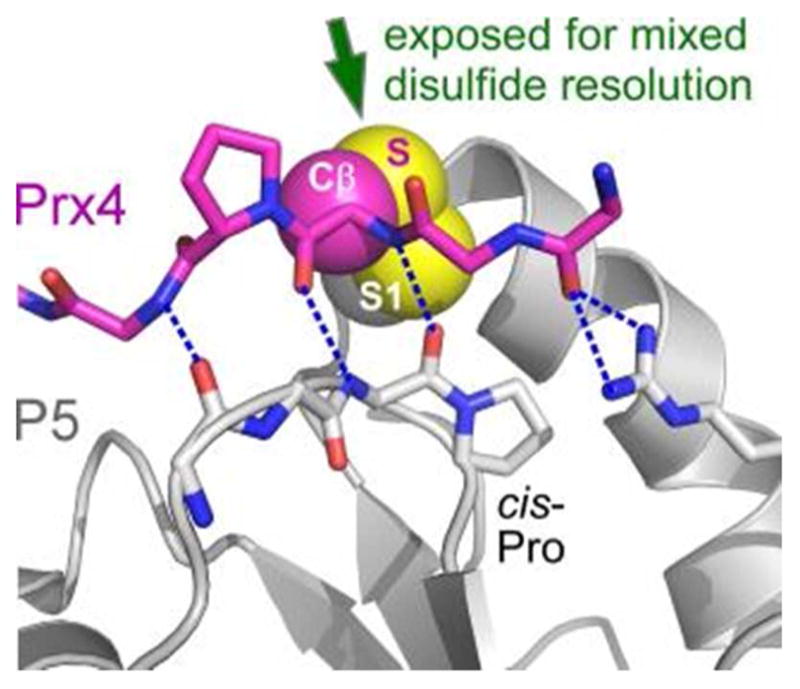
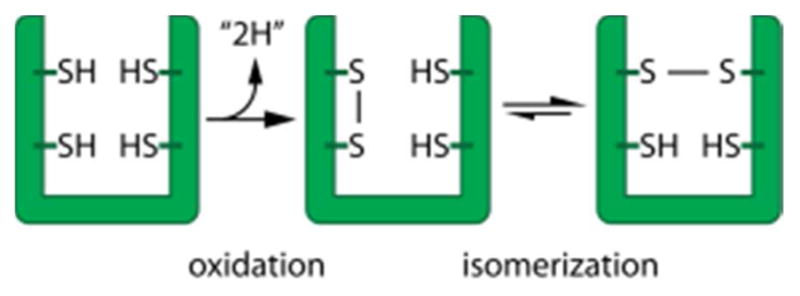
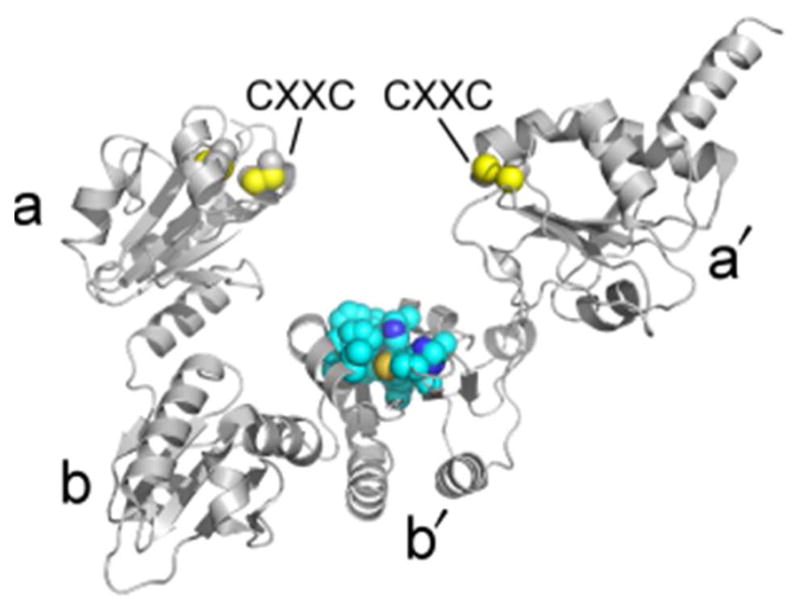


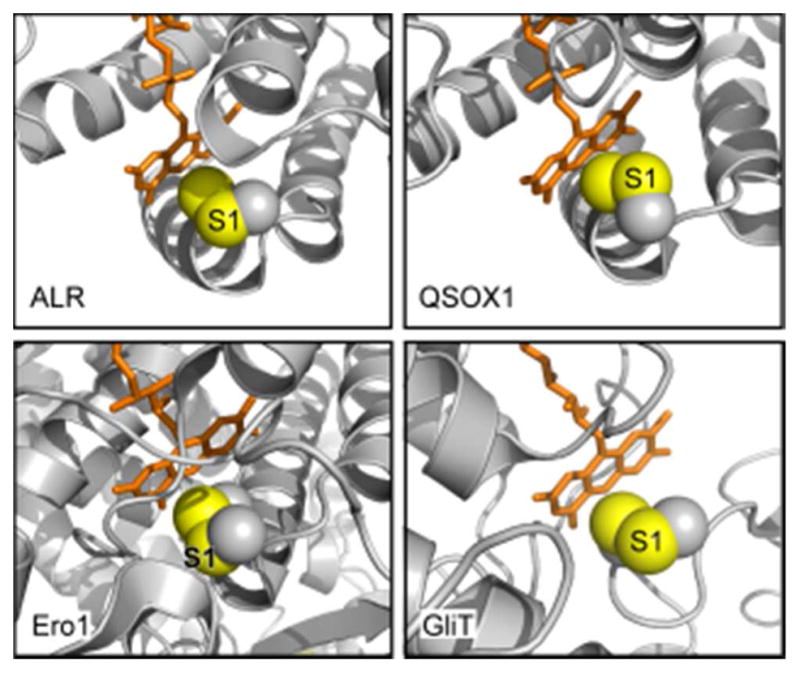
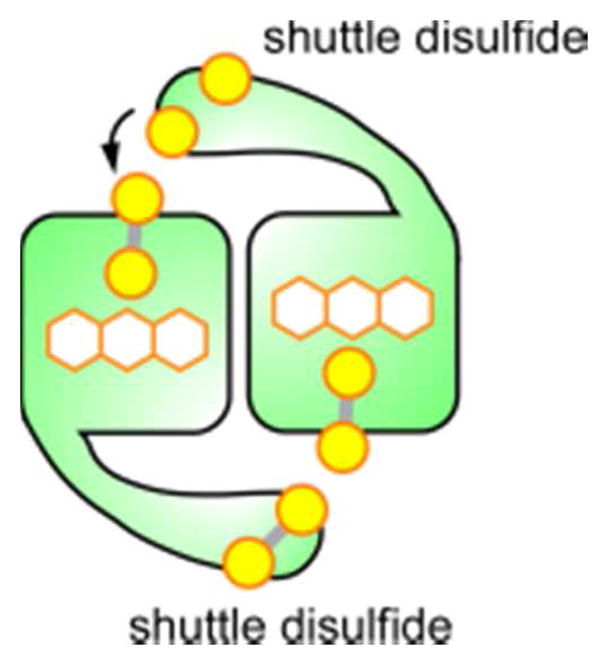
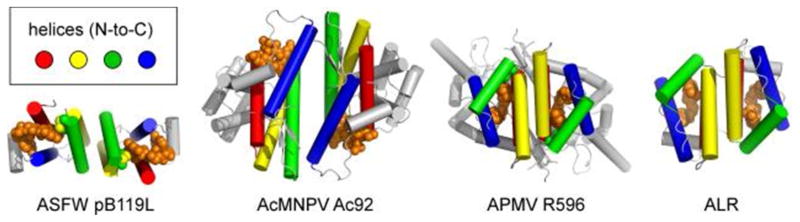
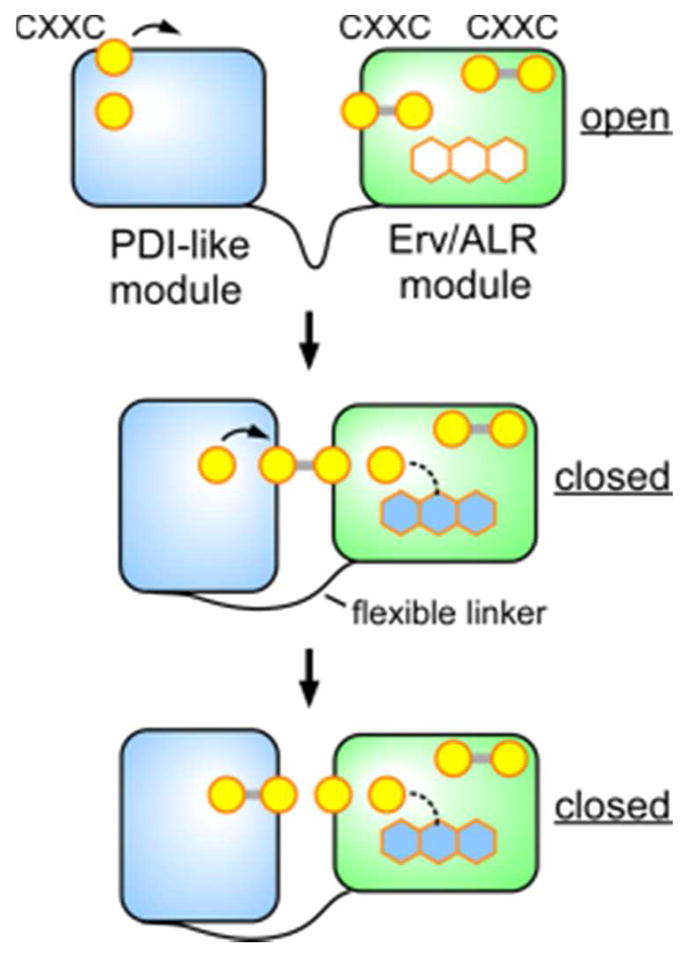
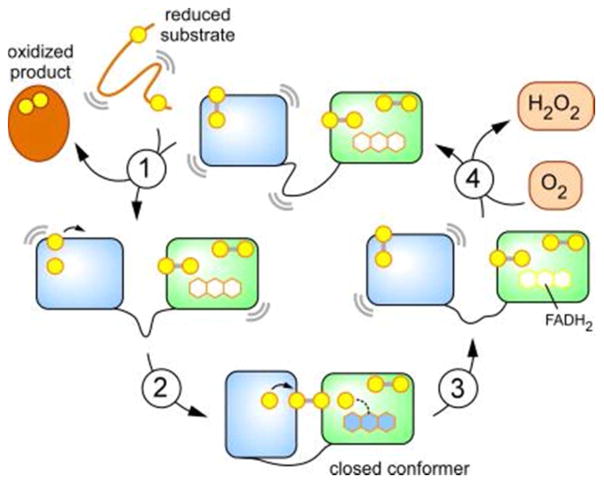
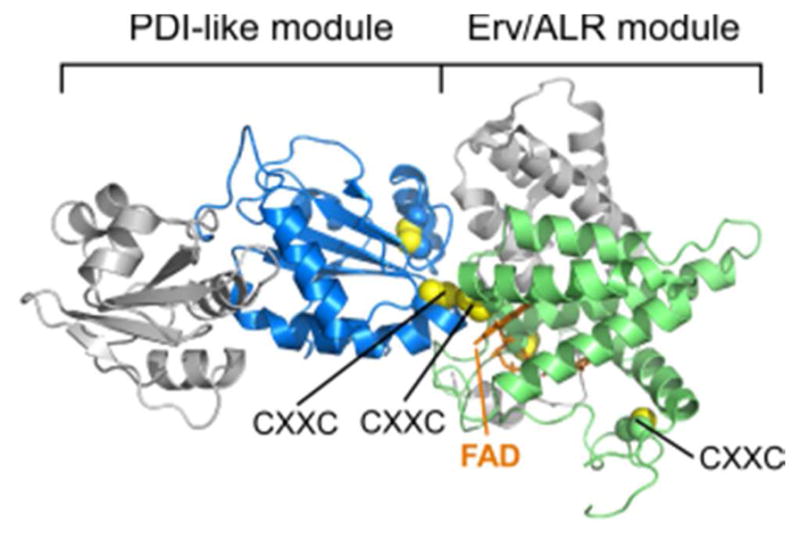

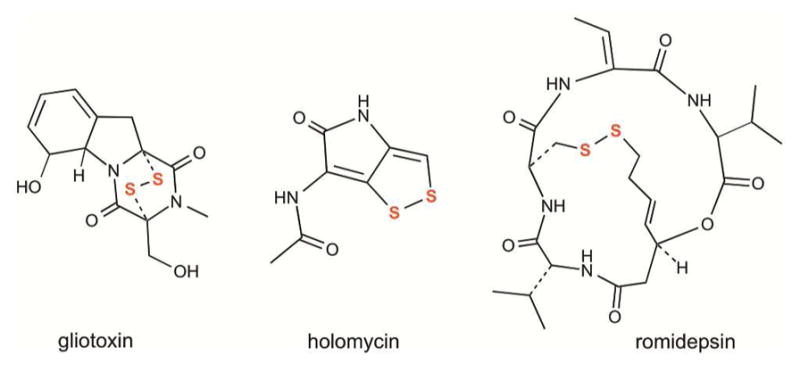
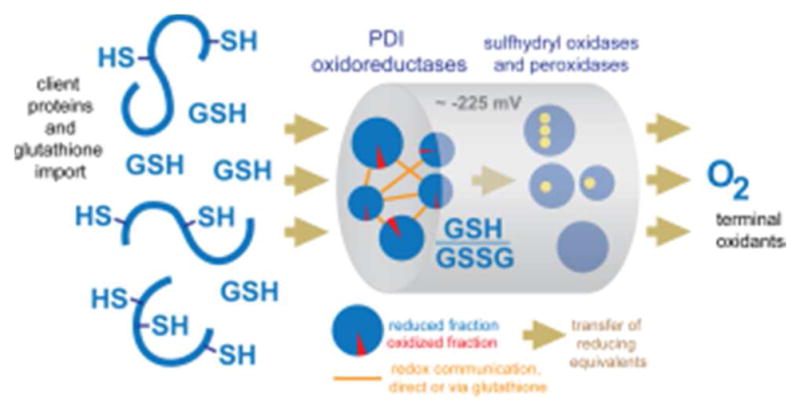
References
-
- Kadokura H, Katzen F, Beckwith J. Protein Disulfide Bond Formation in Prokaryotes. Annu Rev Biochem. 2003;72:111–135. - PubMed
-
- Toth EA, Worby C, Dixon JE, Goedken ER, Marqusee S, Yeates TO. The Crystal Structure of an Adenylosuccinate Lyase from Pyrobaculum aerophilum Reveals an Intracellular Protein with Three Disulfide Bonds. J Mol Biol. 2000;301:433–450. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
