

Viral Activation of Heparanase Drives Pathogenesis of Herpes Simplex Virus-1
- PMID: 28700944
- PMCID: PMC5557421
- DOI: 10.1016/j.celrep.2017.06.041
Viral Activation of Heparanase Drives Pathogenesis of Herpes Simplex Virus-1
Abstract
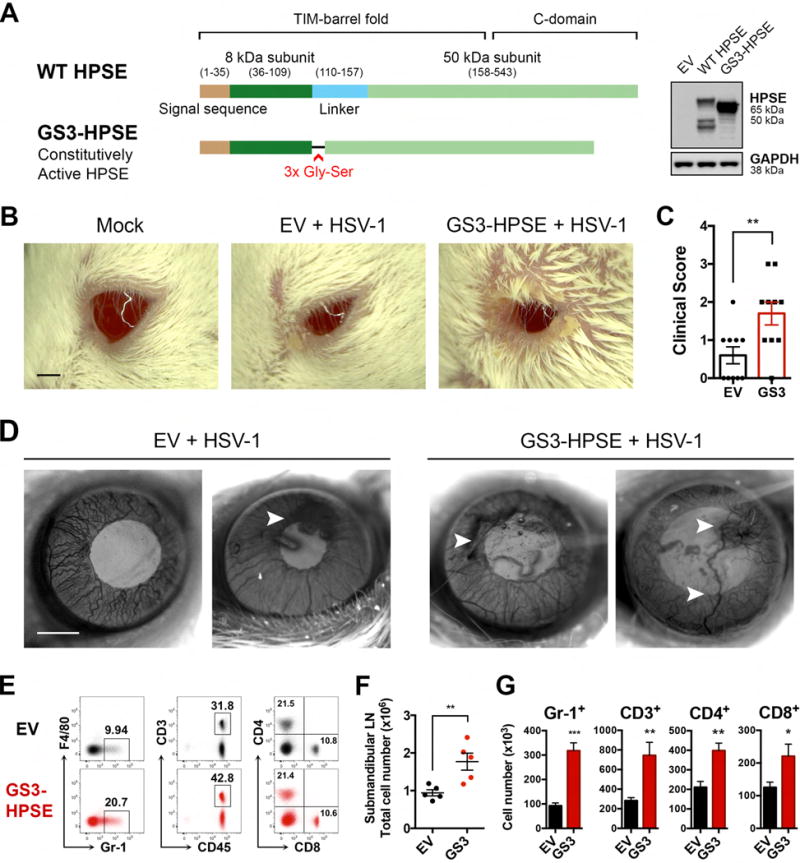
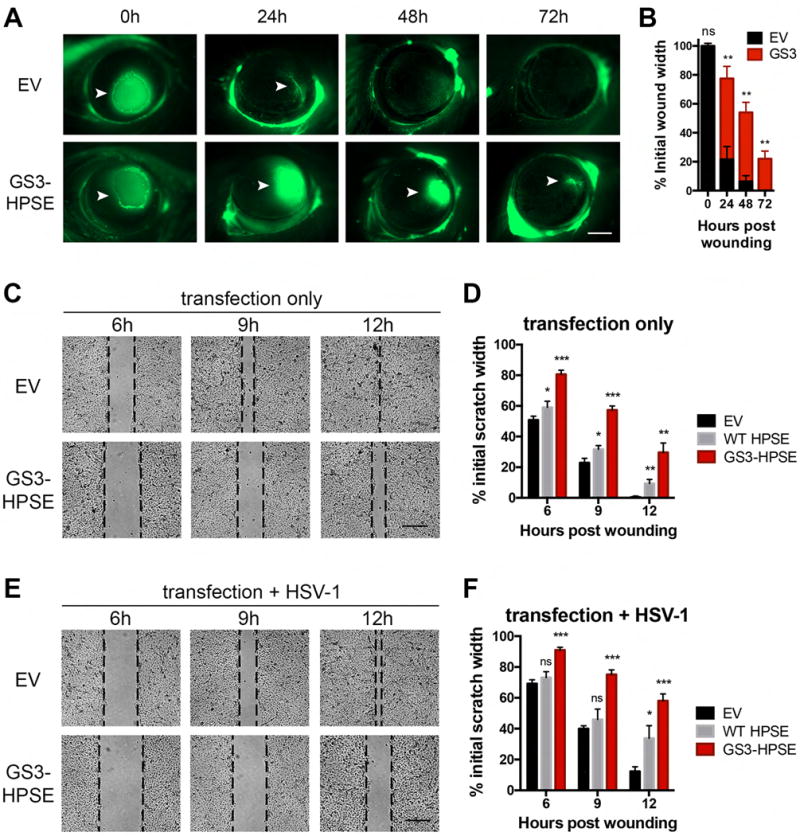
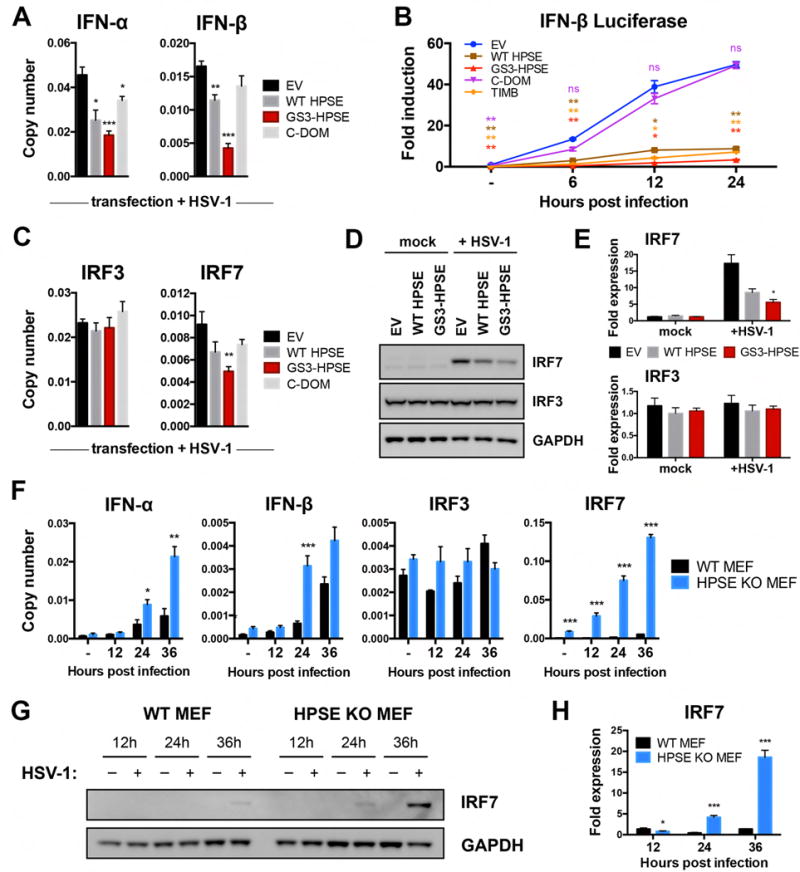
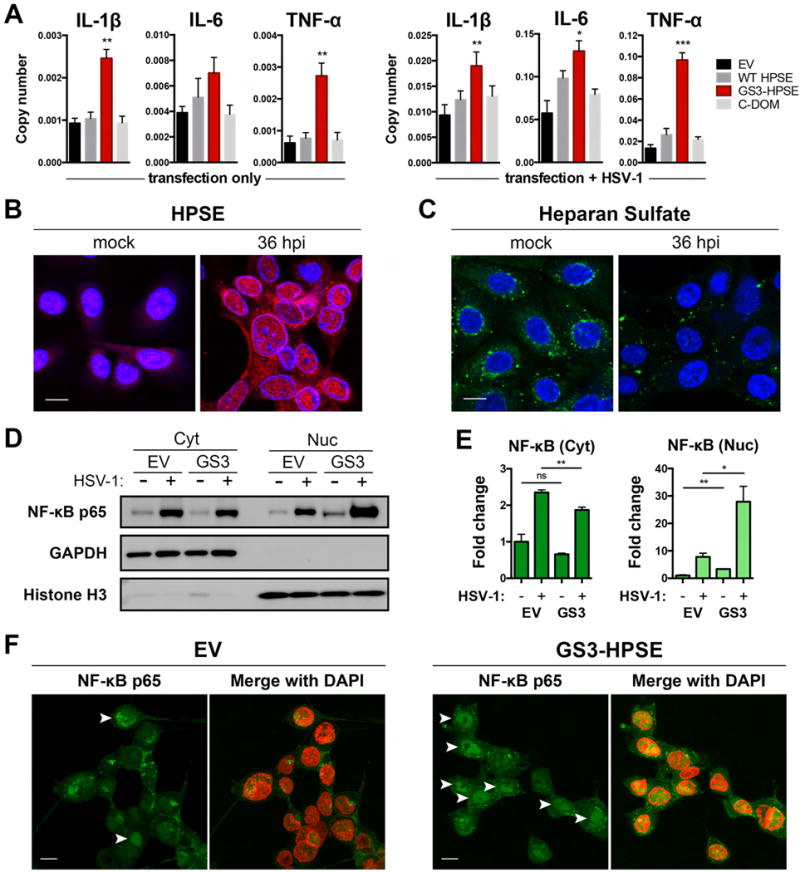
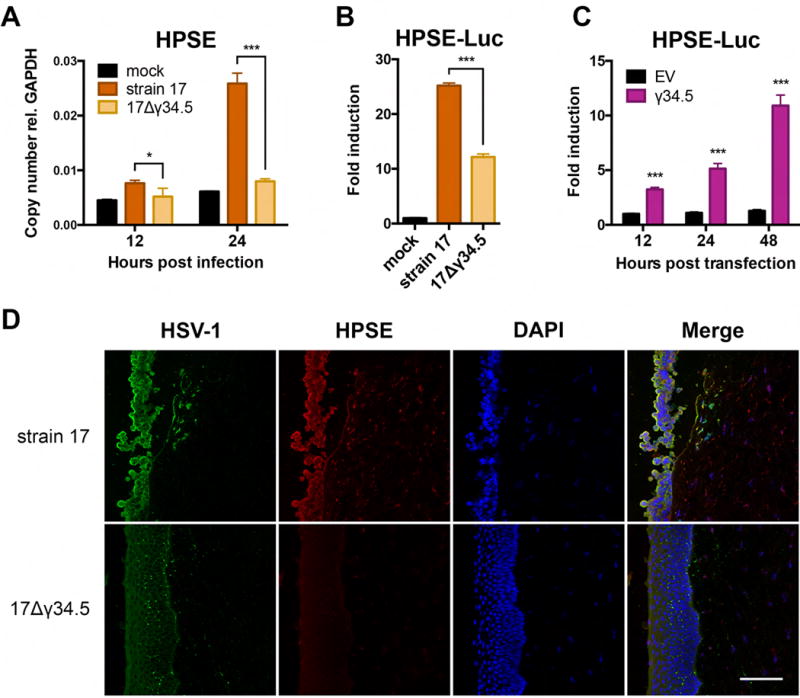
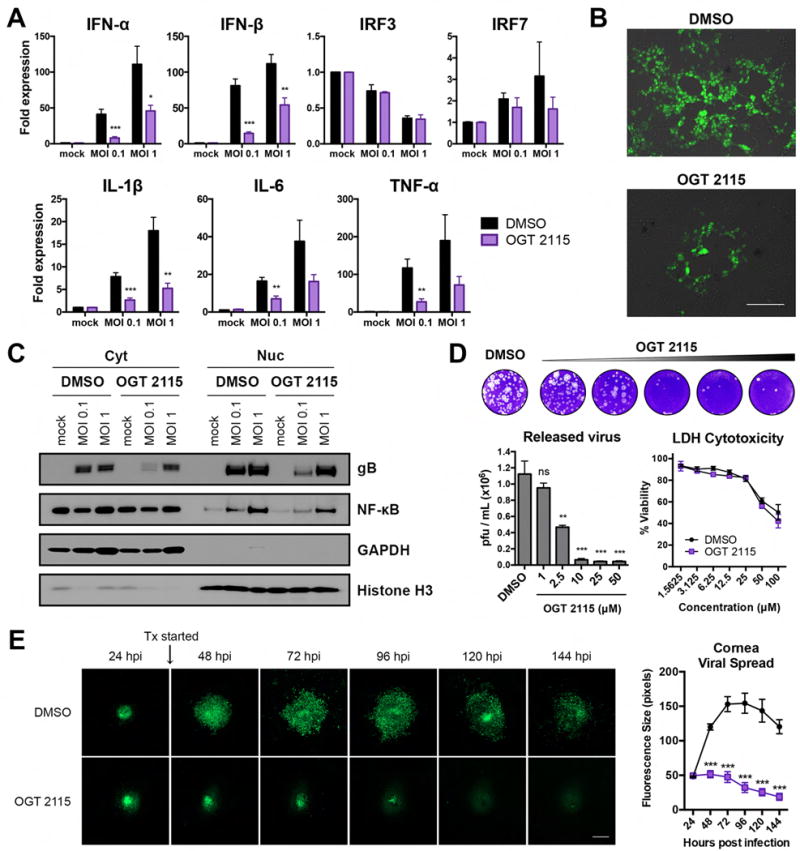
Herpes simplex virus-1 (HSV-1) causes lifelong recurrent pathologies without a cure. How infection by HSV-1 triggers disease processes, especially in the immune-privileged avascular human cornea, remains a major unresolved puzzle. It has been speculated that a cornea-resident molecule must tip the balance in favor of pro-inflammatory and pro-angiogenic conditions observed with herpetic, as well as non-herpetic, ailments of the cornea. Here, we demonstrate that heparanase (HPSE), a host enzyme, is the molecular trigger for multiple pathologies associated with HSV-1 infection. In human corneal epithelial cells, HSV-1 infection upregulates HPSE in a manner dependent on HSV-1 infected cell protein 34.5. HPSE then relocates to the nucleus to regulate cytokine production, inhibits wound closure, enhances viral spread, and thus generates a toxic local environment. Overall, our findings implicate activated HPSE as a driver of viral pathogenesis and call for further attention to this host protein in infection and other inflammatory disorders.
Keywords: cornea; cytokines; heparan sulfate; heparanase; herpes simplex virus; inflammation; interferon; ophthalmology; transcription factors; wound healing.
Copyright © 2017 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
The authors declare that no conflict of interest exists.
Figures
References
-
- Aquino RS, Lee ES, Park PW. Diverse functions of glycosaminoglycans in infectious diseases. Prog Mol Biol Transl Sci. 2010;93:373–394. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
