

Global analysis of protein folding using massively parallel design, synthesis, and testing
- PMID: 28706065
- PMCID: PMC5568797
- DOI: 10.1126/science.aan0693
Global analysis of protein folding using massively parallel design, synthesis, and testing
Abstract
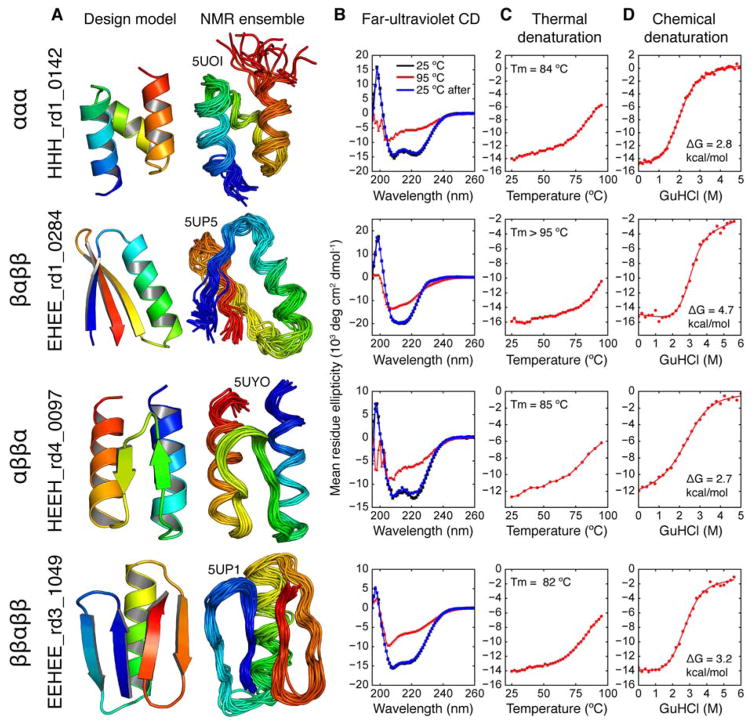
Proteins fold into unique native structures stabilized by thousands of weak interactions that collectively overcome the entropic cost of folding. Although these forces are "encoded" in the thousands of known protein structures, "decoding" them is challenging because of the complexity of natural proteins that have evolved for function, not stability. We combined computational protein design, next-generation gene synthesis, and a high-throughput protease susceptibility assay to measure folding and stability for more than 15,000 de novo designed miniproteins, 1000 natural proteins, 10,000 point mutants, and 30,000 negative control sequences. This analysis identified more than 2500 stable designed proteins in four basic folds-a number sufficient to enable us to systematically examine how sequence determines folding and stability in uncharted protein space. Iteration between design and experiment increased the design success rate from 6% to 47%, produced stable proteins unlike those found in nature for topologies where design was initially unsuccessful, and revealed subtle contributions to stability as designs became increasingly optimized. Our approach achieves the long-standing goal of a tight feedback cycle between computation and experiment and has the potential to transform computational protein design into a data-driven science.
Copyright © 2017, American Association for the Advancement of Science.
Figures




Comment in
-
How do miniproteins fold?Science. 2017 Jul 14;357(6347):133-134. doi: 10.1126/science.aan6864. Science. 2017. PMID: 28706028 No abstract available.
-
Protein design: I like to fold it, fold it.Nat Chem Biol. 2017 Aug 18;13(9):923. doi: 10.1038/nchembio.2467. Nat Chem Biol. 2017. PMID: 28820874 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
