

DNA-PK and P38 MAPK: A Kinase Collusion in Alzheimer's Disease?
- PMID: 28706768
- PMCID: PMC5504707
- DOI: 10.4172/2168-975X.1000232
DNA-PK and P38 MAPK: A Kinase Collusion in Alzheimer's Disease?
Abstract
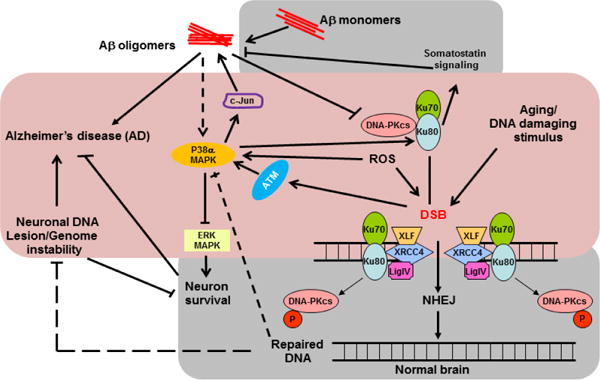
The pathogenesis of Alzheimer's disease (AD), characterized by prevalent neuronal death and extracellular deposit of amyloid plaques, is poorly understood. DNA lesions downstream of reduced DNA repair ability have been reported in AD brains. Neurons predominantly use a mechanism to repair double-strand DNA breaks (DSB), which is non-homologous end joining (NHEJ). NHEJ requires DNA-dependent protein kinase (DNA-PK) activity. DNA-PK is a holoenzyme comprising the p460 kD catalytic subunit (DNA-PKcs) and its activator Ku, a heterodimer of p86 and p70 subunits. Ku first binds and then recruits DNA-PKcs to double-stranded DNA ends before NHEJ process begins. Studies have shown reduced NHEJ activity as well as DNA-PKcs and Ku protein levels in AD brains suggesting possible contribution of unrepaired DSB to AD development. However, normal aging brains also show reduced DNA-PKcs and Ku levels thus challenging the notion of any direct link between NHEJ and AD. Another kinase, p38 MAPK is induced by various DNA damaging agents and DSB itself. Increased DNA damage with aging could induce p38 MAPK and its induction may be sustained when DNA repair is compromised in the brain with reduced DNA-PK activity. Combined, these two events may potentially set the stage for an awry nervous system approaching AD.
Keywords: Amyloid beta; DNA repair; Ku; NHEJ; ROS.
Figures
References
-
- Prince M, Bryce R, Albanese E, Wimo A, Ribeiro W, et al. The global prevalence of dementia: a systematic review and metaanalysis. Alzheimers Dement. 2013;9:63–75 e62. - PubMed
-
- Smith MA, Perry G. The pathogenesis of Alzheimer disease: an alternative to the amyloid hypothesis. J Neuropathol Exp Neurol. 1997;56:217. - PubMed
-
- McKhann GM. Changing concepts of Alzheimer disease. JAMA. 2011;305:2458–2459. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources