

A Missense Mutation in the Extracellular Domain of α ENaC Causes Liddle Syndrome
- PMID: 28710092
- PMCID: PMC5661275
- DOI: 10.1681/ASN.2016111163
A Missense Mutation in the Extracellular Domain of α ENaC Causes Liddle Syndrome
Abstract
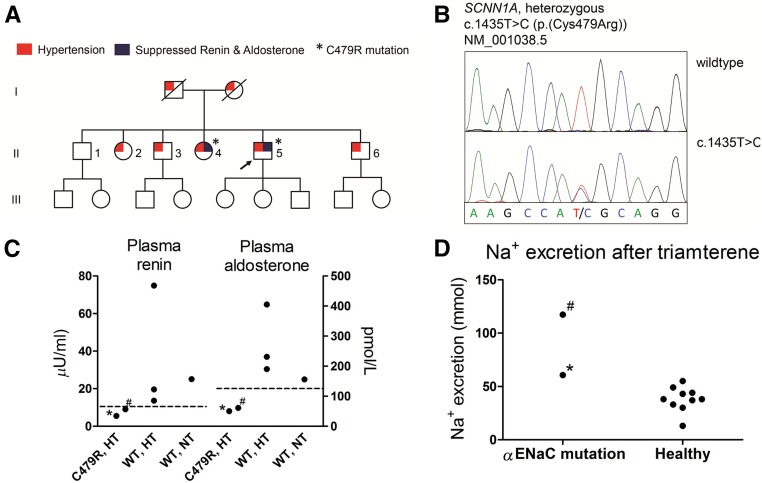
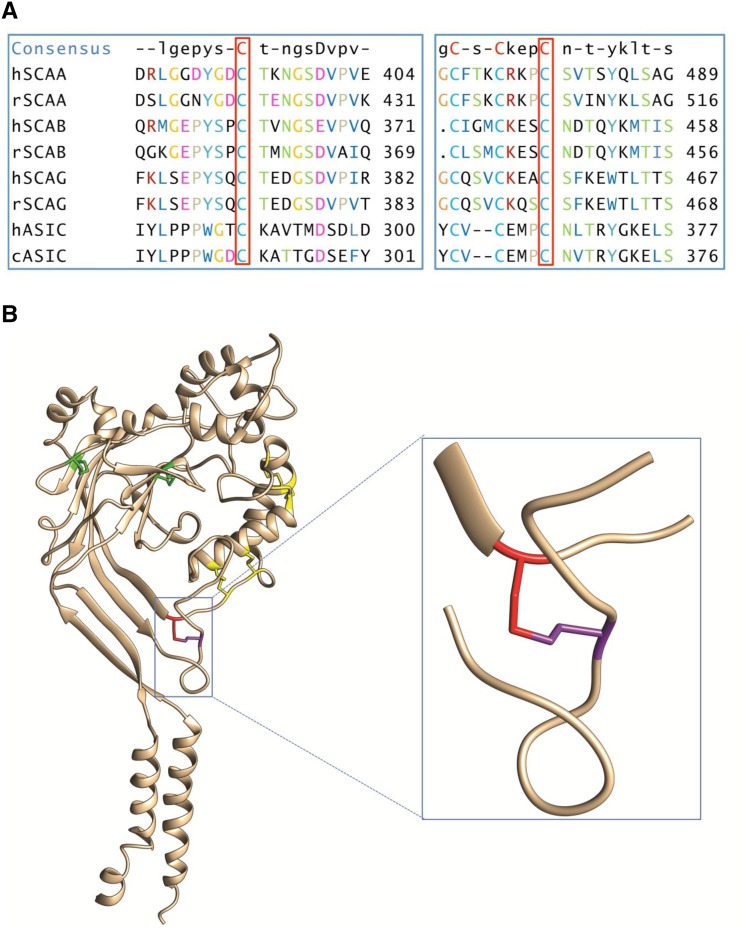
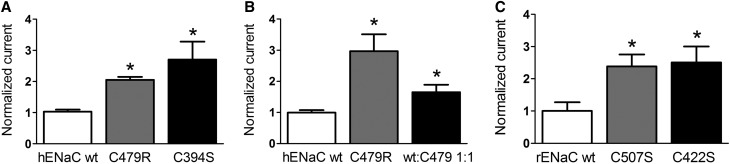
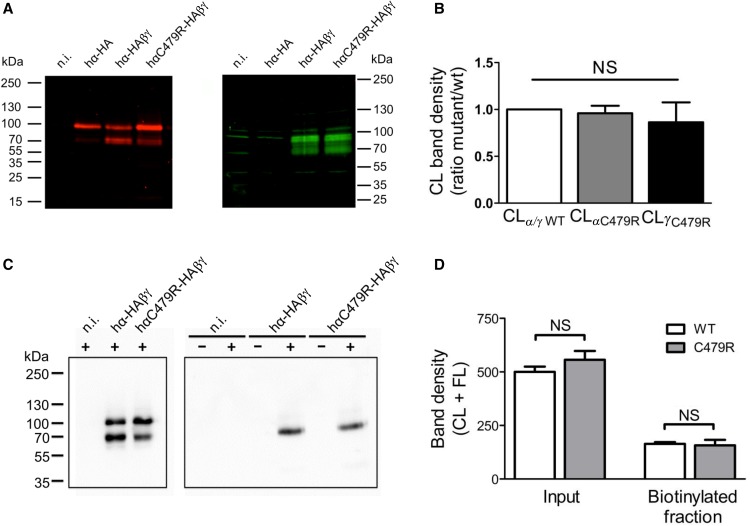
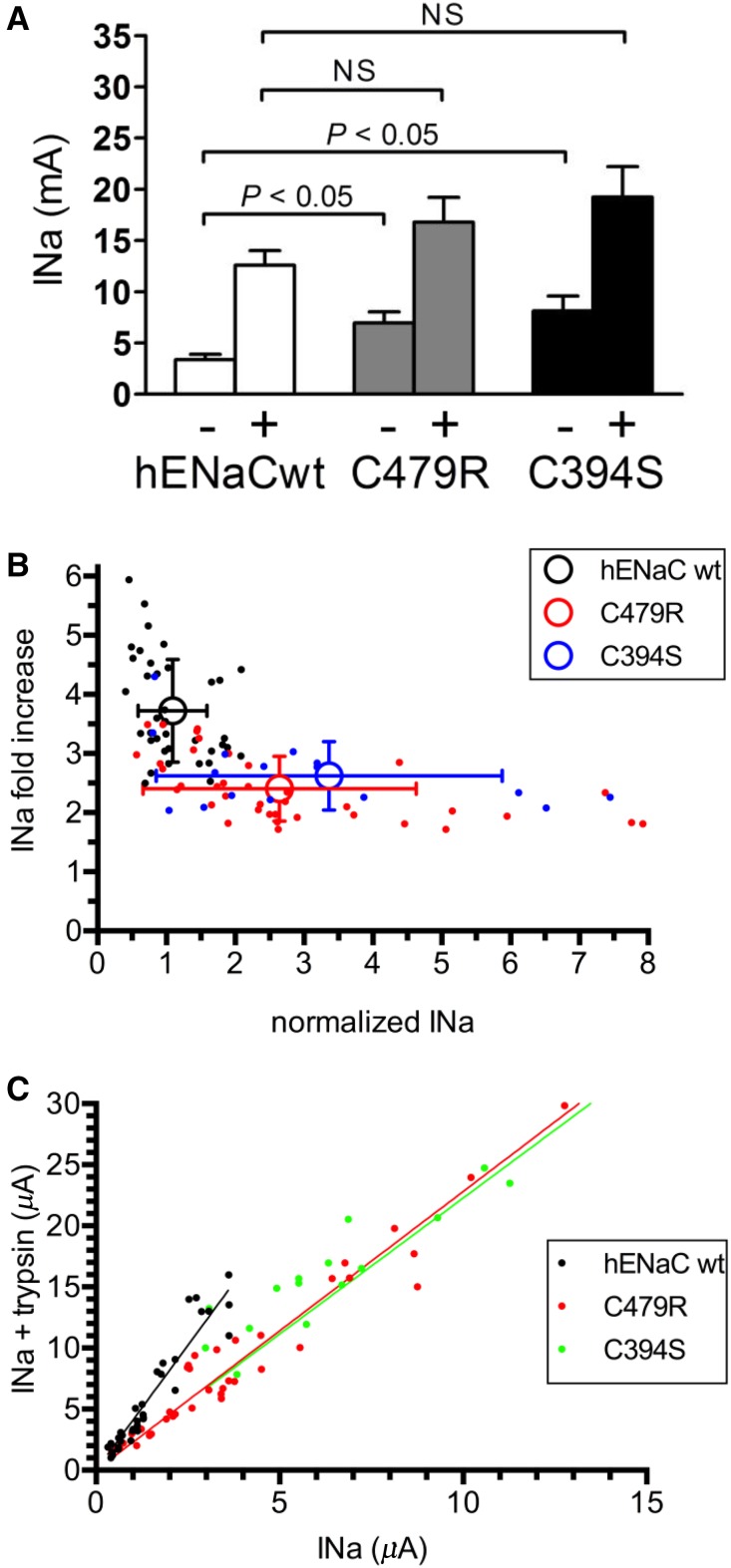
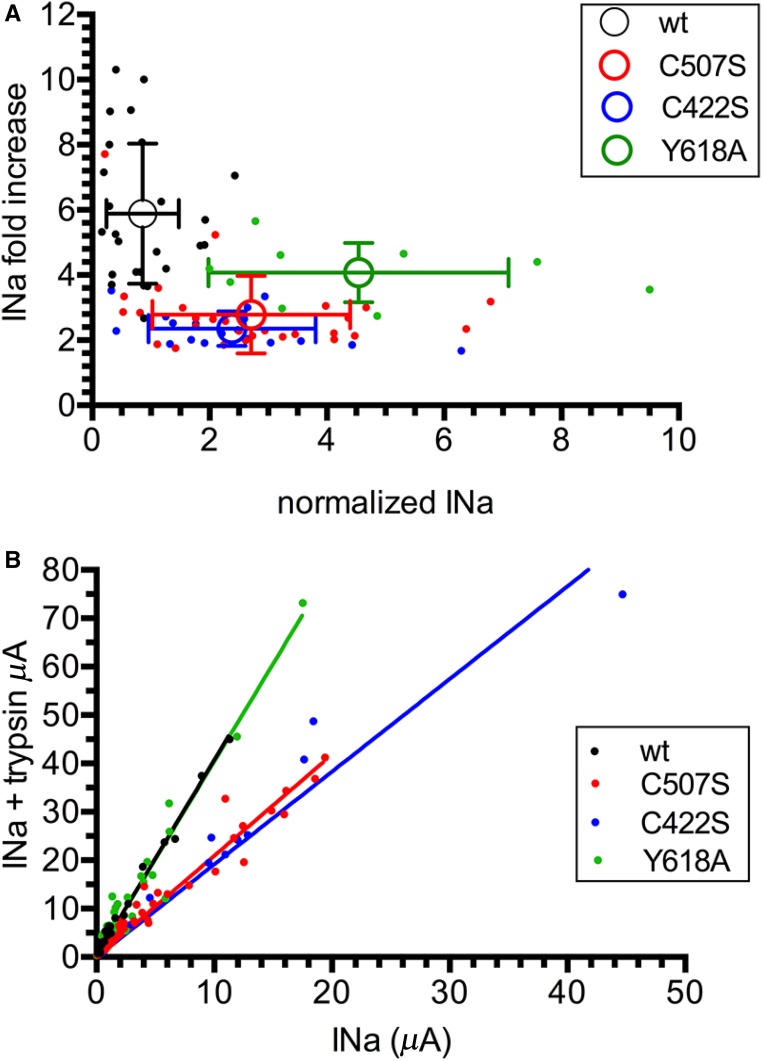
Liddle syndrome is an autosomal dominant form of hypokalemic hypertension due to mutations in the β- or γ-subunit of the epithelial sodium channel (ENaC). Here, we describe a family with Liddle syndrome due to a mutation in αENaC. The proband was referred because of resistant hypokalemic hypertension, suppressed renin and aldosterone, and no mutations in the genes encoding β- or γENaC. Exome sequencing revealed a heterozygous, nonconservative T>C single-nucleotide mutation in αENaC that substituted Cys479 with Arg (C479R). C479 is a highly conserved residue in the extracellular domain of ENaC and likely involved in a disulfide bridge with the partner cysteine C394. In oocytes, the C479R and C394S mutations resulted in similar twofold increases in amiloride-sensitive ENaC current. Quantification of mature cleaved αENaC in membrane fractions showed that the number of channels did not increase with these mutations. Trypsin, which increases open probability of the channel by proteolytic cleavage, resulted in significantly higher currents in the wild type than in C479R or C394S mutants. In summary, a mutation in the extracellular domain of αENaC causes Liddle syndrome by increasing intrinsic channel activity. This mechanism differs from that of the β- and γ-mutations, which result in an increase in channel density at the cell surface. This mutation may explain other cases of patients with resistant hypertension and also provides novel insight into ENaC activation, which is relevant for kidney sodium reabsorption and salt-sensitive hypertension.
Keywords: ENaC; electrophysiology; genetic renal disease; hypertension; hypokalemia.
Copyright © 2017 by the American Society of Nephrology.
Figures
References
-
- Poulter NR, Prabhakaran D, Caulfield M: Hypertension. Lancet 386: 801–812, 2015 - PubMed
-
- Mente A, O’Donnell MJ, Rangarajan S, McQueen MJ, Poirier P, Wielgosz A, Morrison H, Li W, Wang X, Di C, Mony P, Devanath A, Rosengren A, Oguz A, Zatonska K, Yusufali AH, Lopez-Jaramillo P, Avezum A, Ismail N, Lanas F, Puoane T, Diaz R, Kelishadi R, Iqbal R, Yusuf R, Chifamba J, Khatib R, Teo K, Yusuf S; PURE Investigators : Association of urinary sodium and potassium excretion with blood pressure. N Engl J Med 371: 601–611, 2014 - PubMed
-
- Padmanabhan S, Caulfield M, Dominiczak AF: Genetic and molecular aspects of hypertension. Circ Res 116: 937–959, 2015 - PubMed
-
- Lifton RP, Gharavi AG, Geller DS: Molecular mechanisms of human hypertension. Cell 104: 545–556, 2001 - PubMed
-
- Meneton P, Loffing J, Warnock DG: Sodium and potassium handling by the aldosterone-sensitive distal nephron: The pivotal role of the distal and connecting tubule. Am J Physiol Renal Physiol 287: F593–F601, 2004 - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
