

An inhibitor of ubiquitin conjugation and aggresome formation
- PMID: 28717502
- PMCID: PMC5500945
- DOI: 10.1039/c5sc01351h
An inhibitor of ubiquitin conjugation and aggresome formation
Abstract
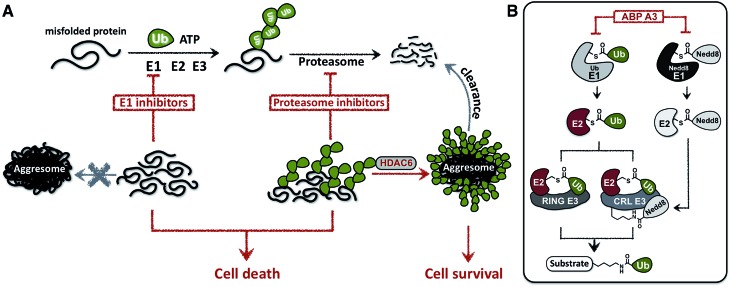
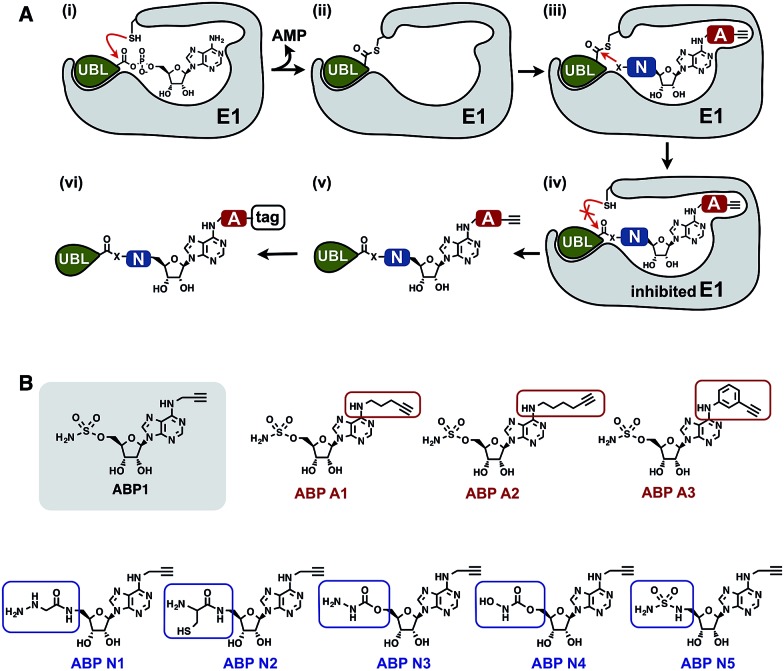
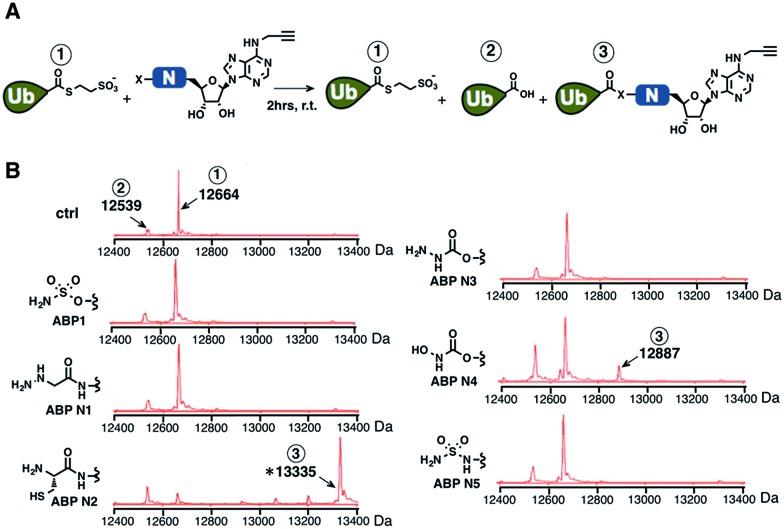
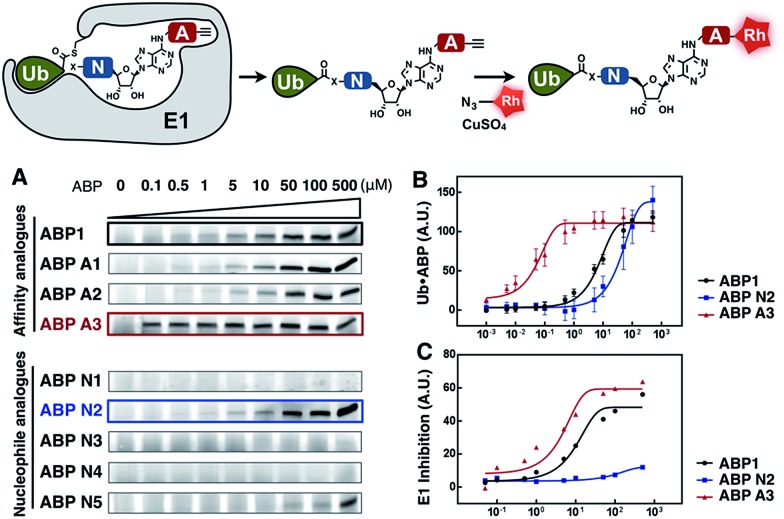
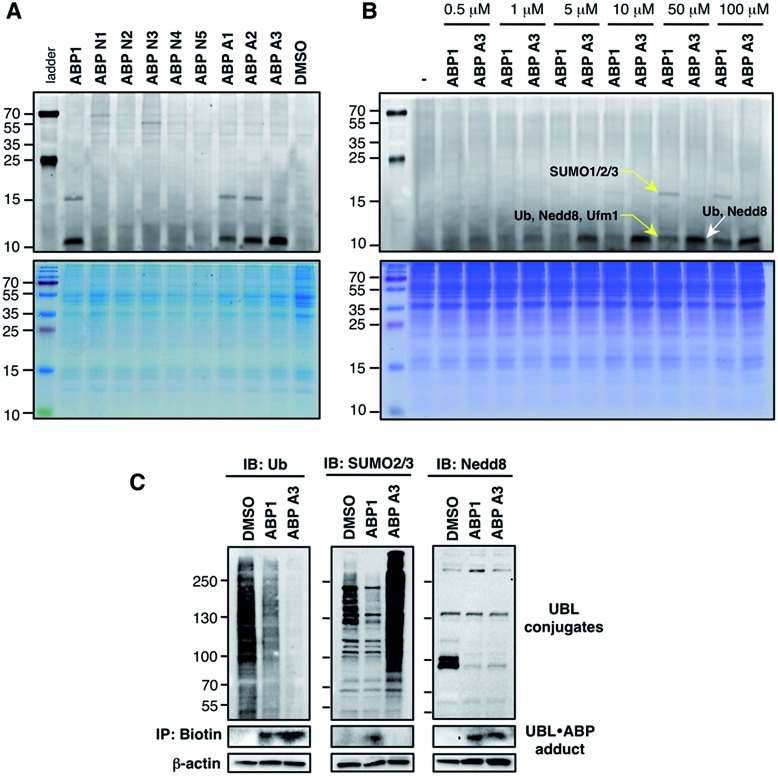
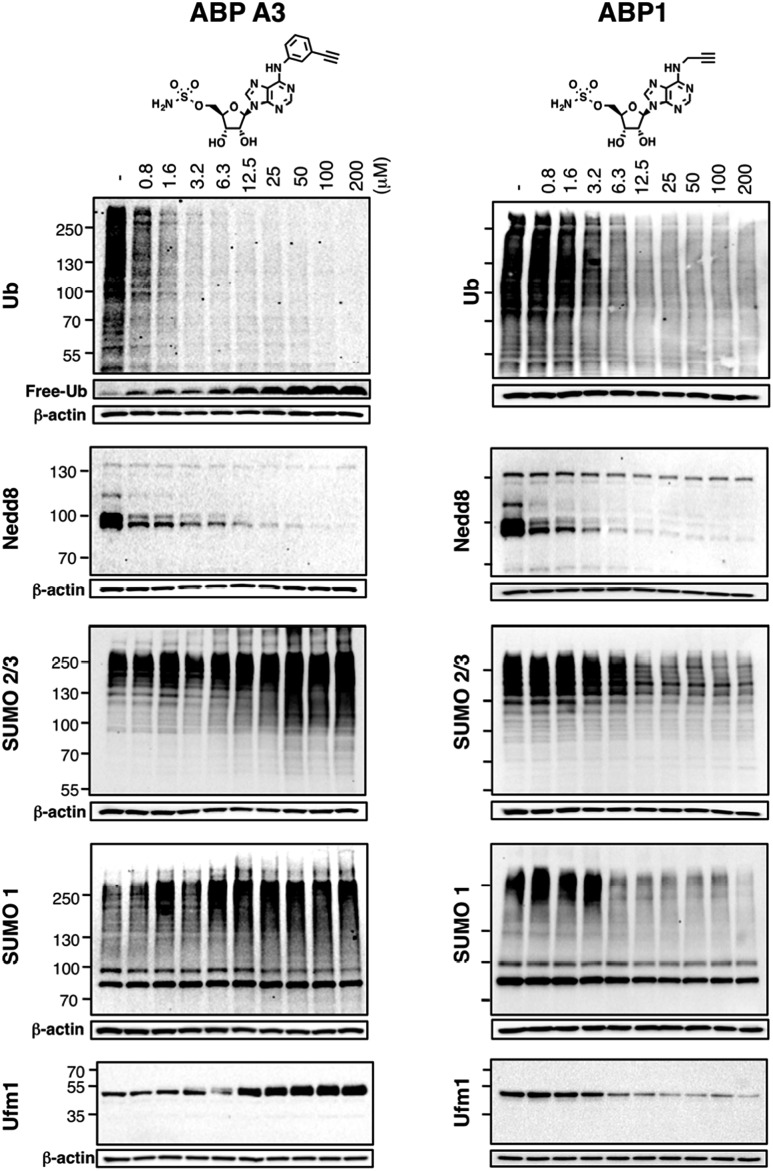
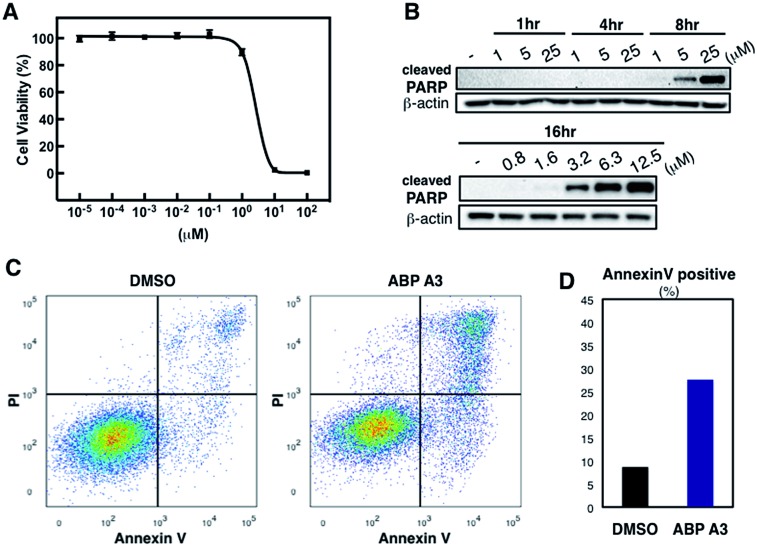
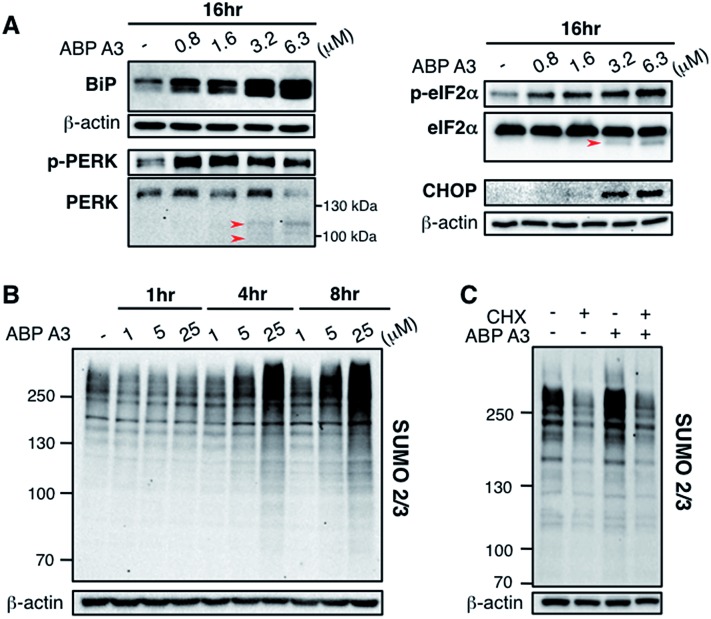
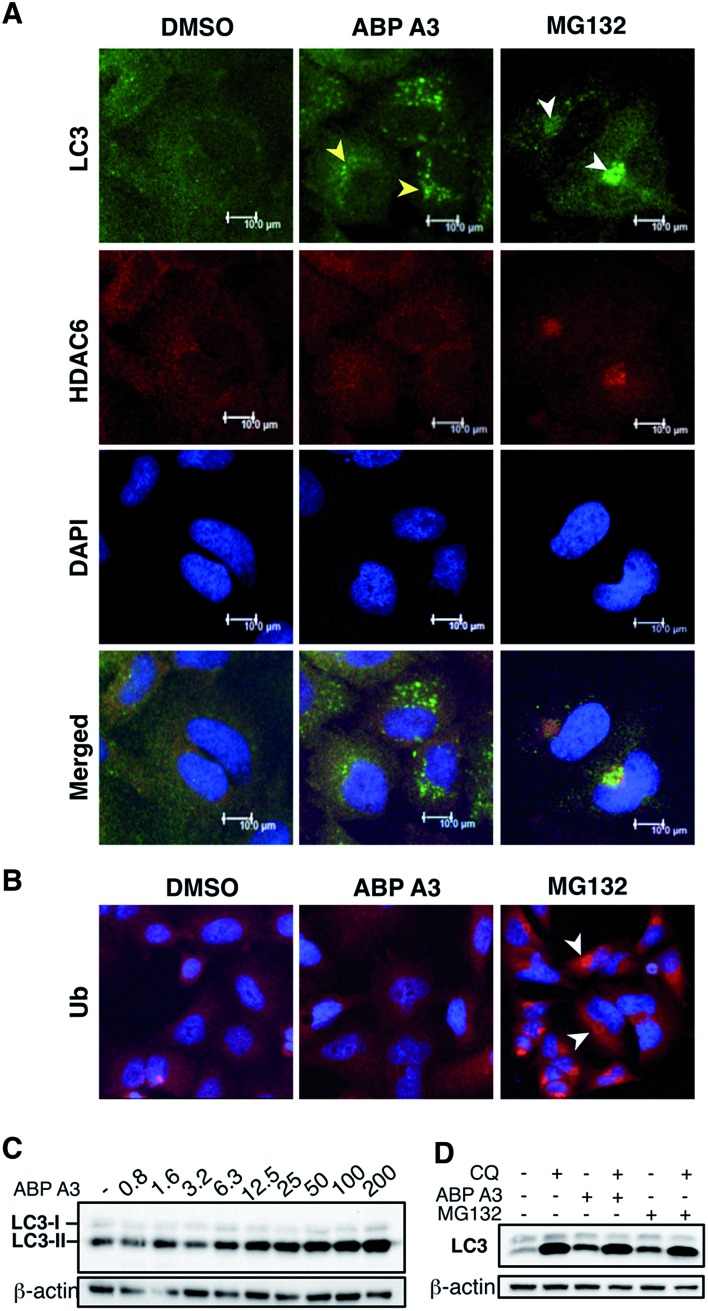
Proteasome inhibitors have revolutionized the treatment of multiple myeloma, and validated the therapeutic potential of the ubiquitin proteasome system (UPS). It is believed that in part, proteasome inhibitors elicit their therapeutic effect by inhibiting the degradation of misfolded proteins, which is proteotoxic and causes cell death. In spite of these successes, proteasome inhibitors are not effective against solid tumors, thus necessitating the need to explore alternative approaches. Furthermore, proteasome inhibitors lead to the formation of aggresomes that clear misfolded proteins via the autophagy-lysosome degradation pathway. Importantly, aggresome formation depends on the presence of polyubiquitin tags on misfolded proteins. We therefore hypothesized that inhibitors of ubiquitin conjugation should inhibit both degradation of misfolded proteins, and ubiquitin dependent aggresome formation, thus outlining the path forward toward more effective anticancer therapeutics. To explore the therapeutic potential of targeting the UPS to treat solid cancers, we have developed an inhibitor of ubiquitin conjugation (ABP A3) that targets ubiquitin and Nedd8 E1 enzymes, enzymes that are required to maintain the activity of the entire ubiquitin system. We have shown that ABP A3 inhibits conjugation of ubiquitin to intracellular proteins and prevents the formation of cytoprotective aggresomes in A549 lung cancer cells. Furthermore, ABP A3 induces activation of the unfolded protein response and apoptosis. Thus, similar to proteasome inhibitors MG132, bortezomib, and carfilzomib, ABP A3 can serve as a novel probe to explore the therapeutic potential of the UPS in solid and hematological malignancies.
Figures
References
-
- Haglund K., Dikic I. J. Cell Sci. 2012;125:265–275. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous