

Clinical and molecular characterization of cystinuria in a French cohort: relevance of assessing large-scale rearrangements and splicing variants
- PMID: 28717662
- PMCID: PMC5511796
- DOI: 10.1002/mgg3.294
Clinical and molecular characterization of cystinuria in a French cohort: relevance of assessing large-scale rearrangements and splicing variants
Abstract
Background: Cystinuria is an autosomal recessive disorder of dibasic amino acid transport in the kidney and the intestine leading to increased urinary cystine excretion and nephrolithiasis. Two genes, SLC3A1 and SLC7A9, coding respectively for rBAT and b0,+AT, account for the genetic basis of cystinuria.
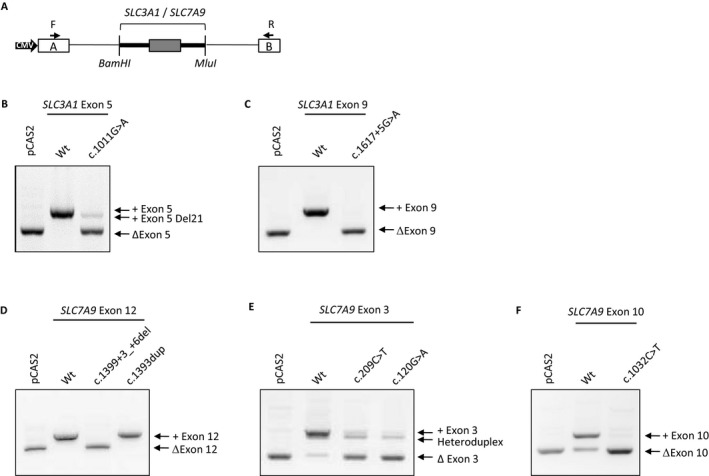
Methods: This study reports the clinical and molecular characterization of a French cohort including 112 cystinuria patients and 25 relatives from 99 families. Molecular screening was performed using sequencing and Quantitative Multiplex PCR of Short Fluorescent Fragments analyses. Functional minigene-based assays have been used to characterize splicing variants.
Results: Eighty-eight pathogenic nucleotide changes were identified in SLC3A1 (63) and SLC7A9 (25) genes, of which 42 were novel. Interestingly, 17% (15/88) and 11% (10/88) of the total number of variants correspond, respectively, to large-scale rearrangements and splicing mutations. Functional minigene-based assays were performed for six variants located outside the most conserved sequences of the splice sites; three variants affect splice sites, while three others modify exonic splicing regulatory elements (ESR), in good agreement with a new in silico prediction based on ΔtESRseq values.
Conclusion: This report expands the spectrum of SLC3A1 and SLC7A9 variants and supports that digenic inheritance is unlikely. Furthermore, it highlights the relevance of assessing large-scale rearrangements and splicing mutations to fully characterize cystinuria patients at the molecular level.
Keywords: Cystinuria; SLC3A1; SLC7A9; exonic splicing regulatory elements; large‐scale rearrangements; splicing mutations.
Figures



References
-
- Albers, A. , Lahme S., Wagner C., Kaiser P., Zerres K., Capasso G., et al. 1999. Mutations in the SLC3A1 gene in cystinuric patients: frequencies and identification of a novel mutation. Genet. Test. 3:227–231. - PubMed
-
- Barbosa, M. , Lopes A., Mota C., Martins E., Oliveira J., Alves S., et al. 2011. Clinical, biochemical and molecular characterization of Cystinuria in a cohort of 12 patients. Clin. Genet. 81:47–55. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
