

Identification of an Alu element-mediated deletion in the promoter region of GNE in siblings with GNE myopathy
- PMID: 28717665
- PMCID: PMC5511805
- DOI: 10.1002/mgg3.300
Identification of an Alu element-mediated deletion in the promoter region of GNE in siblings with GNE myopathy
Abstract
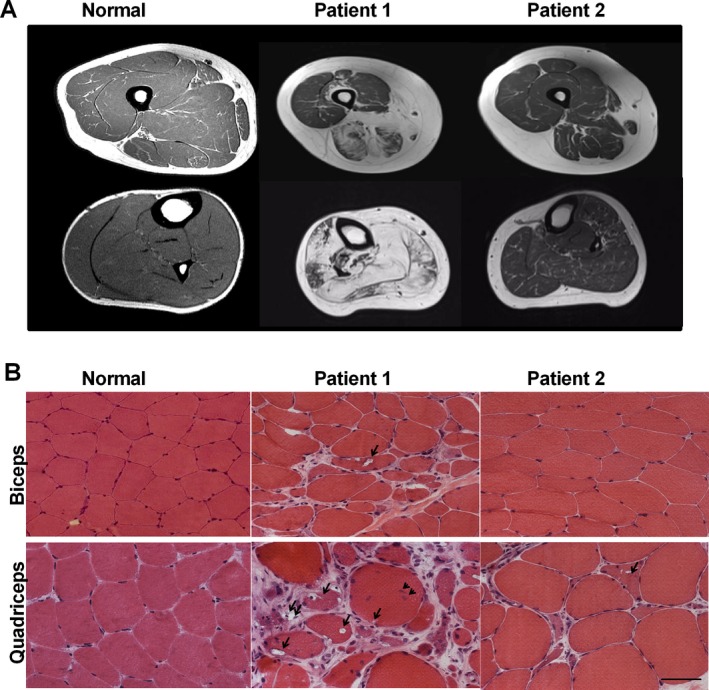
Background: GNE myopathy is a rare genetic disease characterized by progressive muscle atrophy and weakness. It is caused by biallelic mutations in the GNE gene that encodes for the bifunctional enzyme, uridine diphosphate (UDP)-N-acetylglucosamine (GlcNAc) 2-epimerase/N-acetylmannosamine (ManNAc) kinase. Typical characteristics of GNE myopathy include progressive myopathy, first involving anterior tibialis muscle and sparing the quadriceps, and rimmed vacuoles on muscle biopsy. Identifying biallelic mutations by sequencing of the GNE gene confirms the diagnosis of GNE myopathy. In a subset of patients, diagnostic confirmation is challenged by the identification of mutations in only one allele, suggesting mutations in deep intronic regions or regulatory regions.
Methods: We performed targeted sequencing and copy number variant (CNV) analysis of GNE in two siblings who clinically presented with GNE myopathy. Further molecular and biochemical studies were done to characterize the effect of a previously uncharacterized GNE mutation.
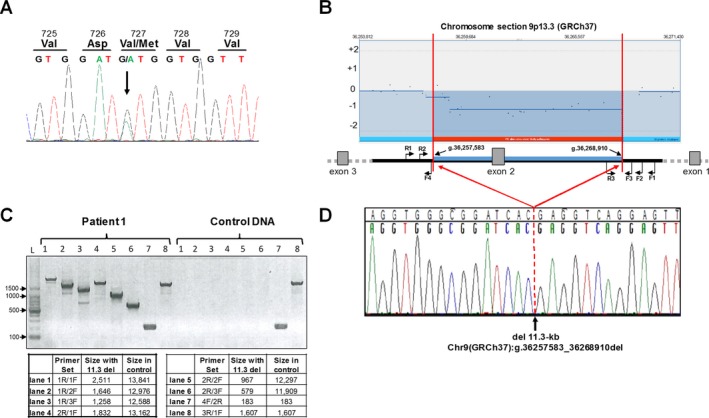
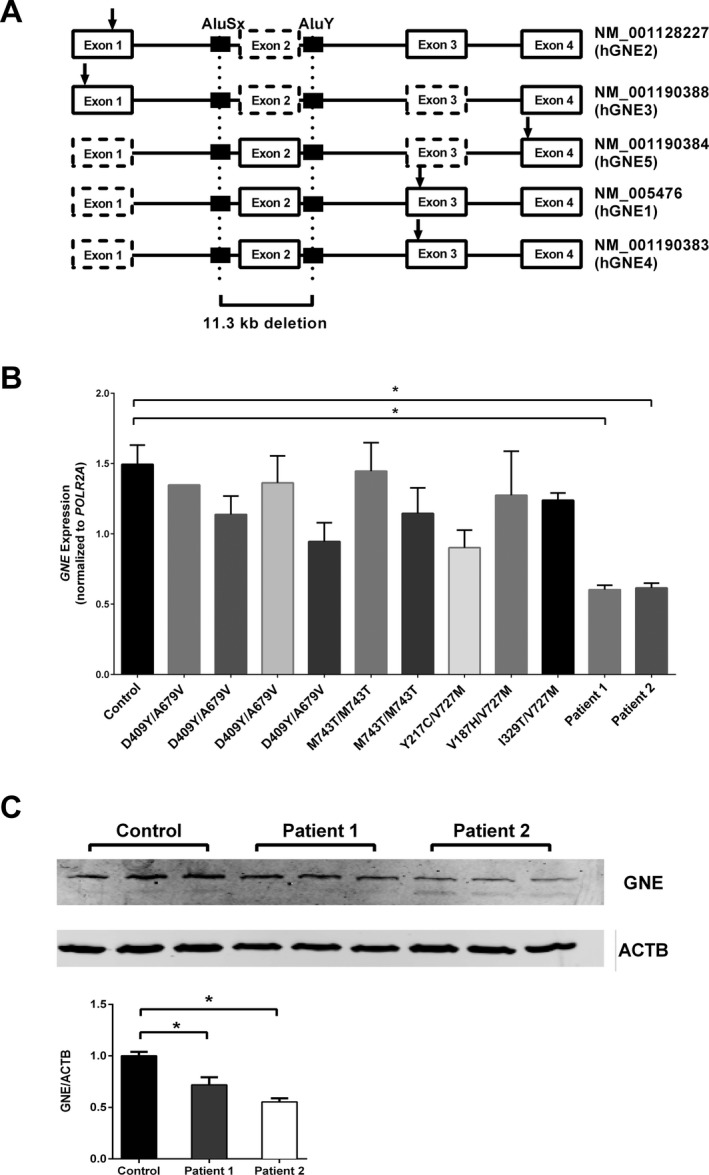
Results: We report two siblings of Indian descent with characteristic features of GNE myopathy, including progressive skeletal muscle weakness initially involving the anterior tibialis, and rimmed vacuoles on muscle biopsy, in which a heterozygous mutation, p.Val727Met, was identified in both affected siblings, but no other deleterious variants in either coding region or exon-intron boundaries of the gene. Subsequent insertion/deletion analysis identified a novel 11.3-kb deletion (Chr9 [GRCh37]: g.36257583_36268910del) encompassing the GNE promoter region, with breakpoints residing in Alu repeats. Gene expression analysis revealed reduced GNE mRNA and protein levels, confirming decreased expression of the deleted allele harboring the deletion.
Conclusions: We have identified GNE as one of the genes susceptible to Alu-mediated recombination. Our findings suggest that the deletion may encompass the promoter or another region necessary for GNE expression. In patients with typical manifestations of GNE myopathy and a single GNE variant identified, copy number variant (CNV) analysis may be useful in arriving at the diagnosis.
Keywords: Alu‐SINE repeat; GNE isoforms; GNE myopathy; array‐CGH; copy number variant; genomic rearrangement; precision medicine; sialic acid.
Figures
References
-
- The National Isometric Muscle Strength (NIMS) Database Consortium 1996. Muscular weakness assessment: use of normal isometric strength data. Arch. Phys. Med. Rehabil. 77:1251–1255. - PubMed
-
- Eisenberg, I. , Avidan N., Potikha T., Hochner H., Chen M., Olender T., et al. 2001. The UDP‐N‐acetylglucosamine 2‐epimerase/N‐acetylmannosamine kinase gene is mutated in recessive hereditary inclusion body myopathy. Nat. Genet. 29:83–87. - PubMed
-
- Ghaderi, D. , Strauss H. M., Reinke S., Cirak S., Reutter W., Lucka L., Hinderlich S.. 2007. Evidence for dynamic interplay of different oligomeric states of UDP‐N‐acetylglucosamine 2‐epimerase/N‐acetylmannosamine kinase by biophysical methods. J. Mol. Biol. 369:746–758. - PubMed
-
- Hinderlich, S. , Stäsche R., Zeitler R., Reutter W.. 1997. A bifunctional enzyme catalyzes the first two steps in N‐acetylneuraminic acid biosynthesis of rat liver. Purification and characterization of UDP‐N‐acetylglucosamine 2‐epimerase/N‐acetylmannosamine kinase. J. Biol. Chem. 272:24313–24318. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
