

Mitotic DNA Damage Response: At the Crossroads of Structural and Numerical Cancer Chromosome Instabilities
- PMID: 28718433
- PMCID: PMC5518619
- DOI: 10.1016/j.trecan.2017.02.001
Mitotic DNA Damage Response: At the Crossroads of Structural and Numerical Cancer Chromosome Instabilities
Abstract
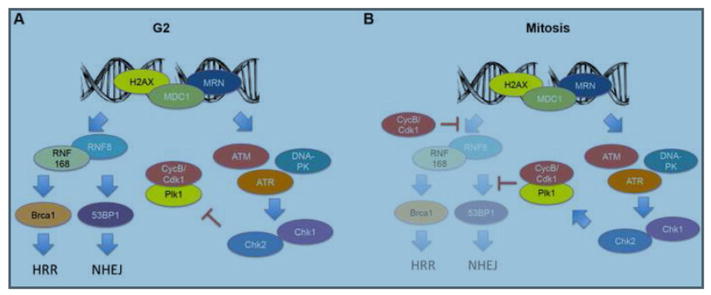
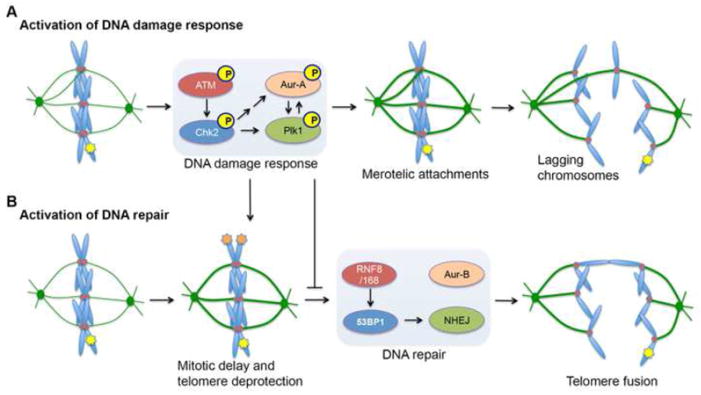
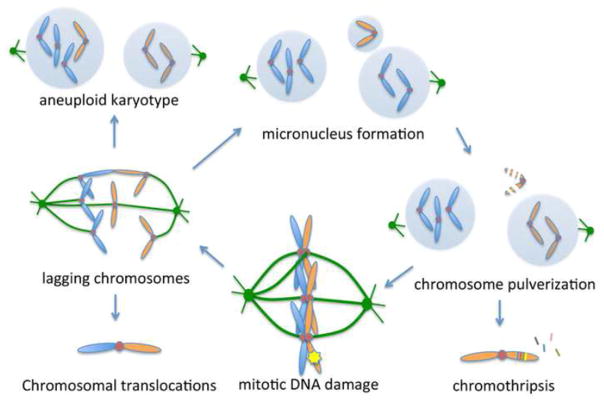
DNA double-strand breaks (DSBs) prevent cells from entering mitosis allowing cells to repair their genomic damage. Little is known about the response to DSBs once cells have already committed to mitosis. Here, we review the genome-protective role of the mitotic DNA damage response (DDR) and evidence suggesting that its untimely activation induces chromosome segregation errors and paradoxically undermines genomic integrity. In contrast to normal cells, cancer cells coopt this pathway to propagate structural and numerical chromosomal instabilities. Cells derived from genomically unstable tumors exhibit evidence for a partially activated DDR during mitosis, which leads to ongoing chromosome segregation errors. Thus, a thorough understanding of the consequences of mitotic DNA damage is key to our ability to devise novel anticancer therapeutic strategies.
Copyright © 2017 Elsevier Inc. All rights reserved.
Conflict of interest statement
Figures
References
-
- Gunderson LL, Tepper JE. Clinical Radiation Oncology. Churchill Livingstone; 2011.
-
- Sinclair WK. Cyclic x-ray responses in mammalian cells in vitro. Radiat Res. 1968;33:620–643. - PubMed
-
- Sinclair WK, Morton RA. X-ray sensitivity during the cell generation cycle of cultured Chinese hamster cells. Radiat Res. 1966;29:450–474. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
