

Interrogating the "unsequenceable" genomic trinucleotide repeat disorders by long-read sequencing
- PMID: 28720120
- PMCID: PMC5514472
- DOI: 10.1186/s13073-017-0456-7
Interrogating the "unsequenceable" genomic trinucleotide repeat disorders by long-read sequencing
Abstract
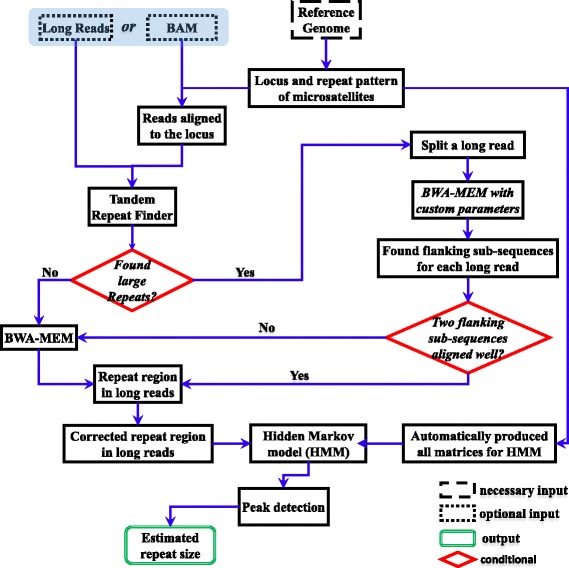
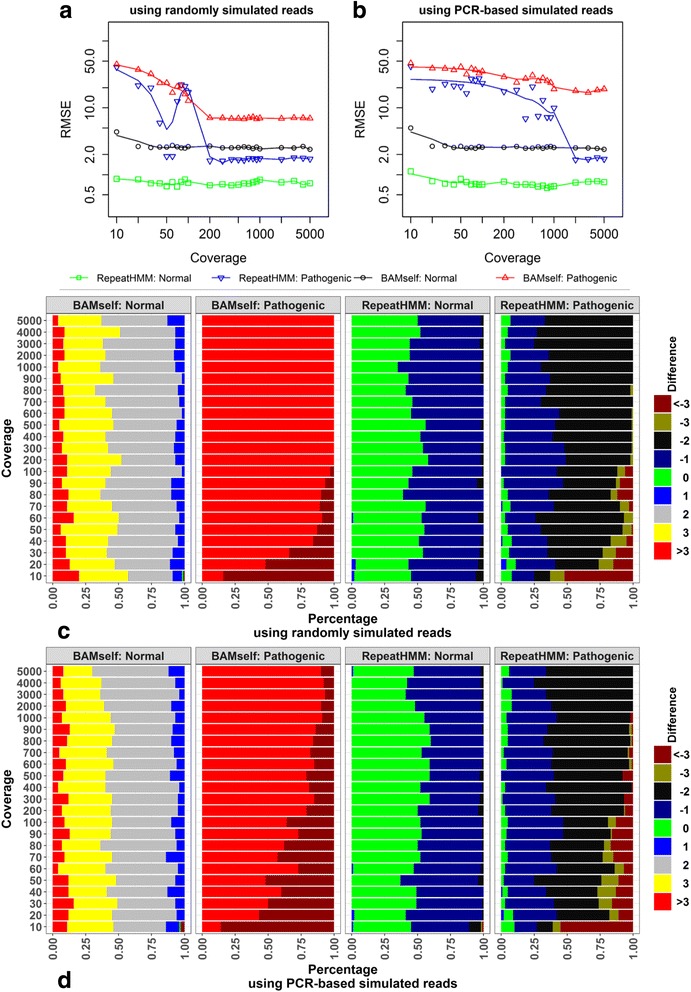
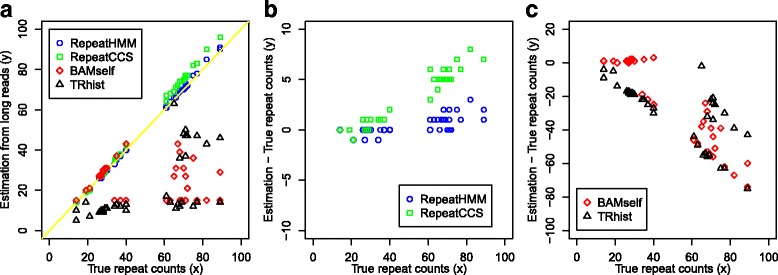
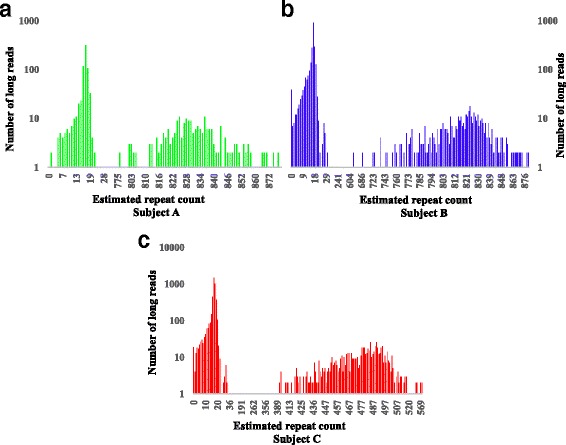
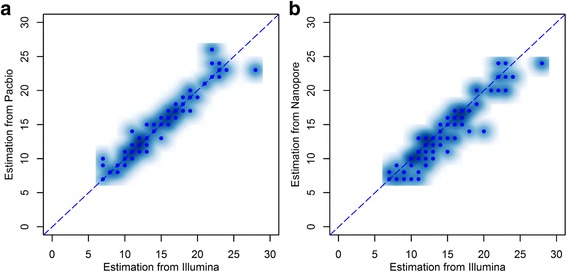
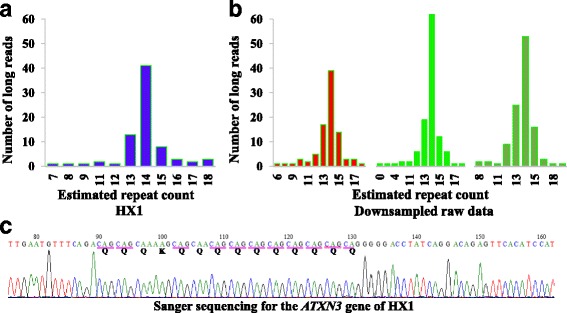
Microsatellite expansion, such as trinucleotide repeat expansion (TRE), is known to cause a number of genetic diseases. Sanger sequencing and next-generation short-read sequencing are unable to interrogate TRE reliably. We developed a novel algorithm called RepeatHMM to estimate repeat counts from long-read sequencing data. Evaluation on simulation data, real amplicon sequencing data on two repeat expansion disorders, and whole-genome sequencing data generated by PacBio and Oxford Nanopore technologies showed superior performance over competing approaches. We concluded that long-read sequencing coupled with RepeatHMM can estimate repeat counts on microsatellites and can interrogate the "unsequenceable" genomic trinucleotide repeat disorders.
Keywords: Long-read sequencing; Microsatellites; Nanopore; PacBio; RepeatHMM; Trinucleotide repeat disorders; Trinucleotide repeats.
Conflict of interest statement
Ethics approval and consent to participate
The study protocol was approved by the China-Japan Friendship Hospital and was conducted in compliance with the Declaration of Helsinki. Written informed consent was obtained from all participants before enrollment.
Consent for publication
All patient information was anonymized at source and unique ID codes were used to identify cases. Publication of de-identified results from all consenting participants was approved.
Competing interests
PZ and DW are employees of Nextomics Bioscences and KW is an advisor for Nextomics Biosciences. All other authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
Similar articles
-
Profiling of Short-Tandem-Repeat Disease Alleles in 12,632 Human Whole Genomes.Am J Hum Genet. 2017 Nov 2;101(5):700-715. doi: 10.1016/j.ajhg.2017.09.013. Am J Hum Genet. 2017. PMID: 29100084 Free PMC article.
-
Genome-wide detection of short tandem repeat expansions by long-read sequencing.BMC Bioinformatics. 2020 Dec 28;21(Suppl 21):542. doi: 10.1186/s12859-020-03876-w. BMC Bioinformatics. 2020. PMID: 33371889 Free PMC article.
-
Approaches to Whole Mitochondrial Genome Sequencing on the Oxford Nanopore MinION.Curr Protoc Hum Genet. 2019 Dec;104(1):e94. doi: 10.1002/cphg.94. Curr Protoc Hum Genet. 2019. PMID: 31743587
-
An update on the neurological short tandem repeat expansion disorders and the emergence of long-read sequencing diagnostics.Acta Neuropathol Commun. 2021 May 25;9(1):98. doi: 10.1186/s40478-021-01201-x. Acta Neuropathol Commun. 2021. PMID: 34034831 Free PMC article. Review.
-
Next-Generation Sequencing and Emerging Technologies.Semin Thromb Hemost. 2019 Oct;45(7):661-673. doi: 10.1055/s-0039-1688446. Epub 2019 May 16. Semin Thromb Hemost. 2019. PMID: 31096307 Review.
Cited by
-
Profiling the Genome-Wide Landscape of Short Tandem Repeats by Long-Read Sequencing.Front Genet. 2022 May 5;13:810595. doi: 10.3389/fgene.2022.810595. eCollection 2022. Front Genet. 2022. PMID: 35601492 Free PMC article.
-
Genetics in mainstream medicine: Finally within grasp to influence healthcare globally.Mol Genet Genomic Med. 2018 May 28;6(4):473-80. doi: 10.1002/mgg3.415. Online ahead of print. Mol Genet Genomic Med. 2018. PMID: 29807392 Free PMC article.
-
Another common genetic ataxia in South Korea: Spinocerebellar ataxia 36.Eur J Hum Genet. 2025 Feb 24. doi: 10.1038/s41431-024-01783-9. Online ahead of print. Eur J Hum Genet. 2025. PMID: 39994402
-
Structural variation in the sequencing era.Nat Rev Genet. 2020 Mar;21(3):171-189. doi: 10.1038/s41576-019-0180-9. Epub 2019 Nov 15. Nat Rev Genet. 2020. PMID: 31729472 Free PMC article. Review.
-
NanoVar: accurate characterization of patients' genomic structural variants using low-depth nanopore sequencing.Genome Biol. 2020 Mar 3;21(1):56. doi: 10.1186/s13059-020-01968-7. Genome Biol. 2020. PMID: 32127024 Free PMC article.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
