

The challenges and promise of targeting the Liver X Receptors for treatment of inflammatory disease
- PMID: 28720427
- PMCID: PMC5743771
- DOI: 10.1016/j.pharmthera.2017.07.010
The challenges and promise of targeting the Liver X Receptors for treatment of inflammatory disease
Abstract
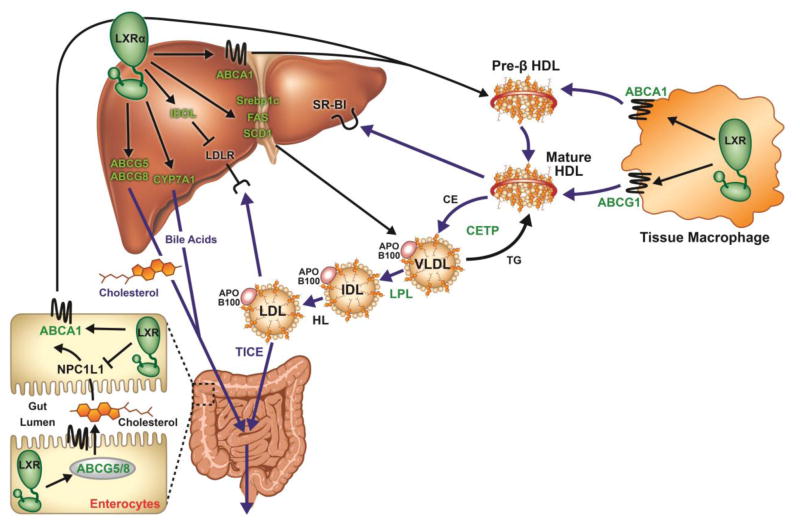
The Liver X Receptors (LXRs) are oxysterol-activated transcription factors that upregulate a suite of genes that together promote coordinated mobilization of excess cholesterol from cells and from the body. The LXRs, like other nuclear receptors, are anti-inflammatory, inhibiting signal-dependent induction of pro-inflammatory genes by nuclear factor-κB, activating protein-1, and other transcription factors. Synthetic LXR agonists have been shown to ameliorate atherosclerosis and a wide range of inflammatory disorders in preclinical animal models. Although this has suggested potential for application to human disease, systemic LXR activation is complicated by hepatic steatosis and hypertriglyceridemia, consequences of lipogenic gene induction in the liver by LXRα. The past several years have seen the development of multiple advanced LXR therapeutics aiming to avoid hepatic lipogenesis, including LXRβ-selective agonists, tissue-selective agonists, and transrepression-selective agonists. Although several synthetic LXR agonists have made it to phase I clinical trials, none have progressed due to unforeseen adverse reactions or undisclosed reasons. Nonetheless, several sophisticated pharmacologic strategies, including structure-guided drug design, cell-specific drug targeting, as well as non-systemic drug routes have been initiated and remain to be comprehensively explored. In addition, recent studies have identified potential utility for targeting the LXRs during therapy with other agents, such as glucocorticoids and rexinoids. Despite the pitfalls encountered to date in translation of LXR agonists to human disease, it appears likely that this accelerating field will ultimately yield effective and safe applications for LXR targeting in humans.
Keywords: Atherosclerosis; Cholesterol; Inflammation; Liver X Receptor; Oxysterol; Reverse cholesterol transport.
Published by Elsevier Inc.
Conflict of interest statement
Figures

References
-
- A-Gonzalez N, Bensinger SJ, Hong C, Beceiro S, Bradley MN, Zelcer N, Deniz J, Ramirez C, Diaz M, Gallardo G, de Galarreta CR, Salazar J, Lopez F, Edwards P, Parks J, Andujar M, Tontonoz P, Castrillo A. Apoptotic cells promote their own clearance and immune tolerance through activation of the nuclear receptor LXR. Immunity. 2009;31:245–258. - PMC - PubMed
-
- A-Gonzalez N, Guillen JA, Gallardo G, Diaz M, de la Rosa JV, Hernandez IH, Casanova-Acebes M, Lopez F, Tabraue C, Beceiro S, Hong C, Lara PC, Andujar M, Arai S, Miyazaki T, Li S, Corbi AL, Tontonoz P, Hidalgo A, Castrillo A. The nuclear receptor LXRalpha controls the functional specialization of splenic macrophages. Nat Immunol. 2013;14:831–839. - PMC - PubMed
-
- Asquith DL, Miller AM, Hueber AJ, McKinnon HJ, Sattar N, Graham GJ, McInnes IB. Liver X receptor agonism promotes articular inflammation in murine collagen-induced arthritis. Arthritis Rheum. 2009;60:2655–2665. - PubMed
-
- Asquith DL, Miller AM, Reilly J, Kerr S, Welsh P, Sattar N, McInnes IB. Simultaneous activation of the liver X receptors (LXRalpha and LXRbeta) drives murine collagen-induced arthritis disease pathology. Ann Rheum Dis. 2011;70:2225–2228. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
