

Potential for Monitoring Gut Microbiota for Diagnosing Infections and Graft-versus-Host Disease in Cancer and Stem Cell Transplant Patients
- PMID: 28720679
- PMCID: PMC6053668
- DOI: 10.1373/clinchem.2016.259499
Potential for Monitoring Gut Microbiota for Diagnosing Infections and Graft-versus-Host Disease in Cancer and Stem Cell Transplant Patients
Abstract
Background: Gut microbiota, the collective community of microorganisms inhabiting the intestine, have been shown to provide many beneficial functions for the host. Recent advances in next-generation sequencing and advanced molecular biology approaches have allowed researchers to identify gut microbiota signatures associated with disease processes and, in some cases, establish causality and elucidate underlying mechanisms.
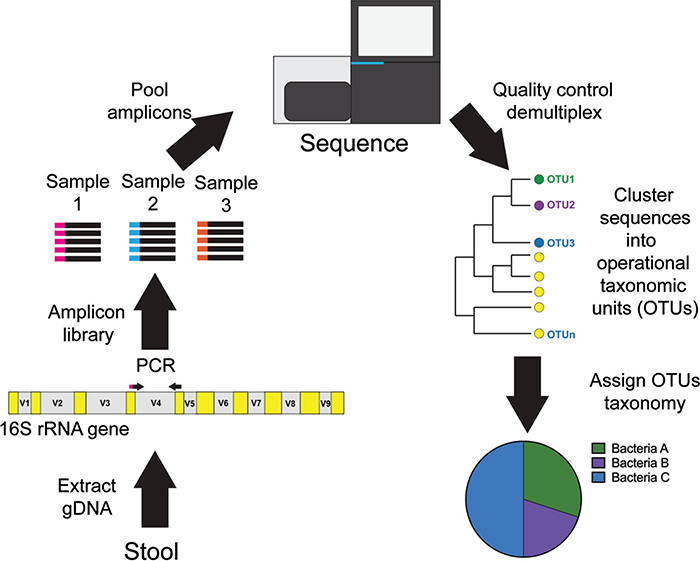
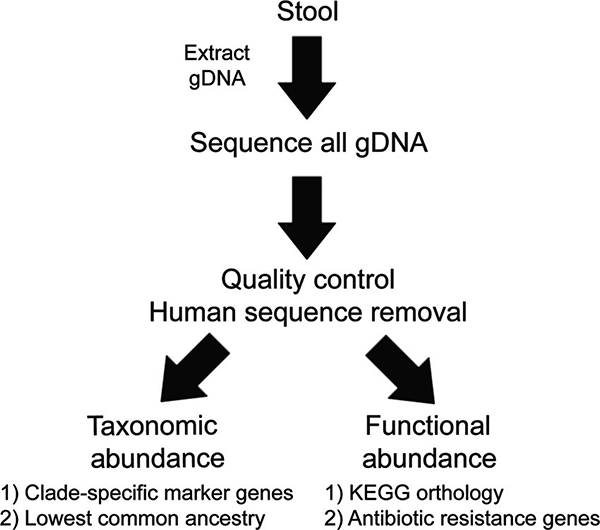
Content: This report reviews 3 commonly used methods for studying the gut microbiota and microbiome (the collective genomes of the gut microorganisms): 16S rRNA gene sequencing, bacterial group or species-specific quantitative polymerase chain reaction (qPCR), and metagenomic shotgun sequencing (MSS). The technical approaches and resources needed for each approach are outlined, and advantages and disadvantages for each approach are summarized. The findings regarding the role of the gut microbiota in the health of patients with cancer and stem cell transplant (SCT) patients (specifically in modulating the development of gut-derived bacterial infections and a posttransplant immune-mediated complication known as graft-vs-host-disease) are reviewed. Finally, there is discussion of the potential viability of these approaches in the actual clinical treatment of cancer and SCT patients.
Summary: Advances in next-generation sequencing have revolutionized our understanding of the importance of the gut microbiome to human health. Both 16S rRNA gene sequencing and MSS are currently too labor-intensive or computationally burdensome to incorporate into real-time clinical monitoring of gut microbiomes. Yet, the lessons learned from these technologies could be adapted to currently used methods (e.g., qPCR) that could then be rigorously tested in the clinical care of these patients.
© 2017 American Association for Clinical Chemistry.
Conflict of interest statement
Figures

References
-
- Spor A, Koren O, Ley R. Unravelling the effects of the environment and host genotype on the gut microbiome. Nat Rev Microbiol 2011;9:279–90. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
