

Hitchhiking and epistasis give rise to cohort dynamics in adapting populations
- PMID: 28720700
- PMCID: PMC5547604
- DOI: 10.1073/pnas.1702314114
Hitchhiking and epistasis give rise to cohort dynamics in adapting populations
Abstract
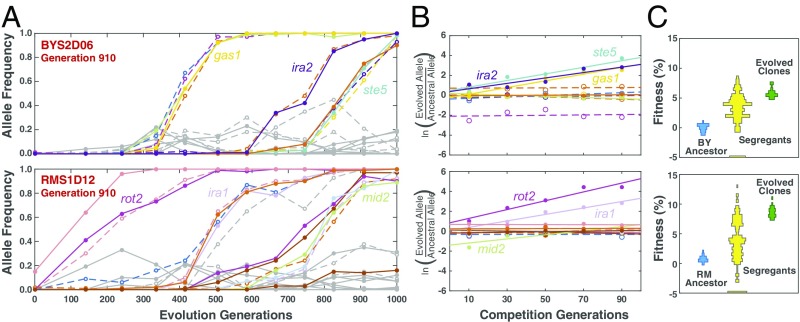
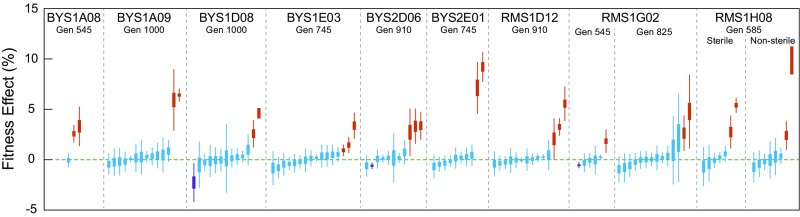
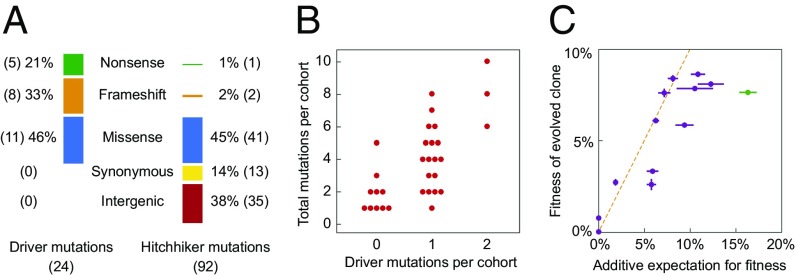
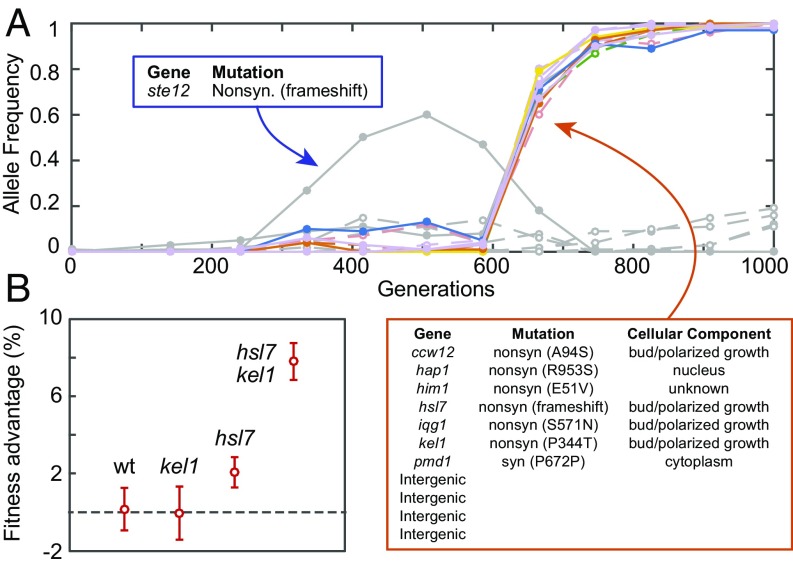
Beneficial mutations are the driving force of adaptive evolution. In asexual populations, the identification of beneficial alleles is confounded by the presence of genetically linked hitchhiker mutations. Parallel evolution experiments enable the recognition of common targets of selection; yet these targets are inherently enriched for genes of large target size and mutations of large effect. A comprehensive study of individual mutations is necessary to create a realistic picture of the evolutionarily significant spectrum of beneficial mutations. Here we use a bulk-segregant approach to identify the beneficial mutations across 11 lineages of experimentally evolved yeast populations. We report that nearly 80% of detected mutations have no discernible effects on fitness and less than 1% are deleterious. We determine the distribution of driver and hitchhiker mutations in 31 mutational cohorts, groups of mutations that arise synchronously from low frequency and track tightly with one another. Surprisingly, we find that one-third of cohorts lack identifiable driver mutations. In addition, we identify intracohort synergistic epistasis between alleles of hsl7 and kel1, which arose together in a low-frequency lineage.
Keywords: cohorts; epistasis; experimental evolution; fitness.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
