

Comparative integrated omics: identification of key functionalities in microbial community-wide metabolic networks
- PMID: 28721231
- PMCID: PMC5515219
- DOI: 10.1038/npjbiofilms.2015.7
Comparative integrated omics: identification of key functionalities in microbial community-wide metabolic networks
Abstract
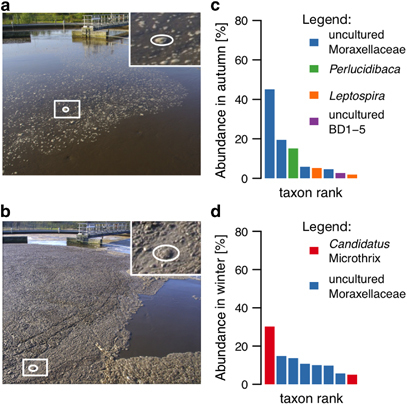
Background: Mixed microbial communities underpin important biotechnological processes such as biological wastewater treatment (BWWT). A detailed knowledge of community structure and function relationships is essential for ultimately driving these systems towards desired outcomes, e.g., the enrichment in organisms capable of accumulating valuable resources during BWWT.
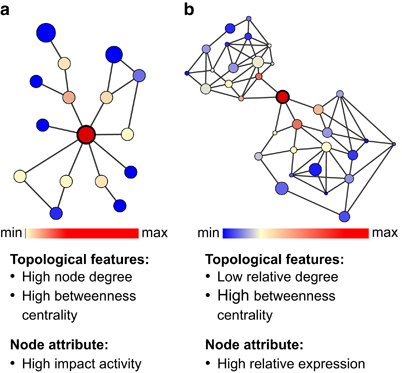
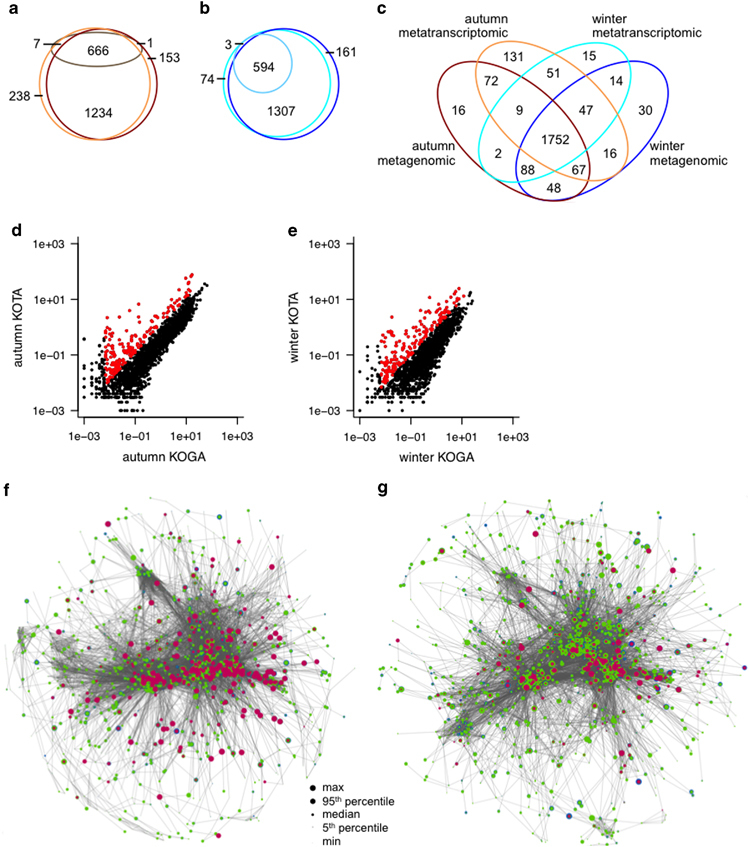
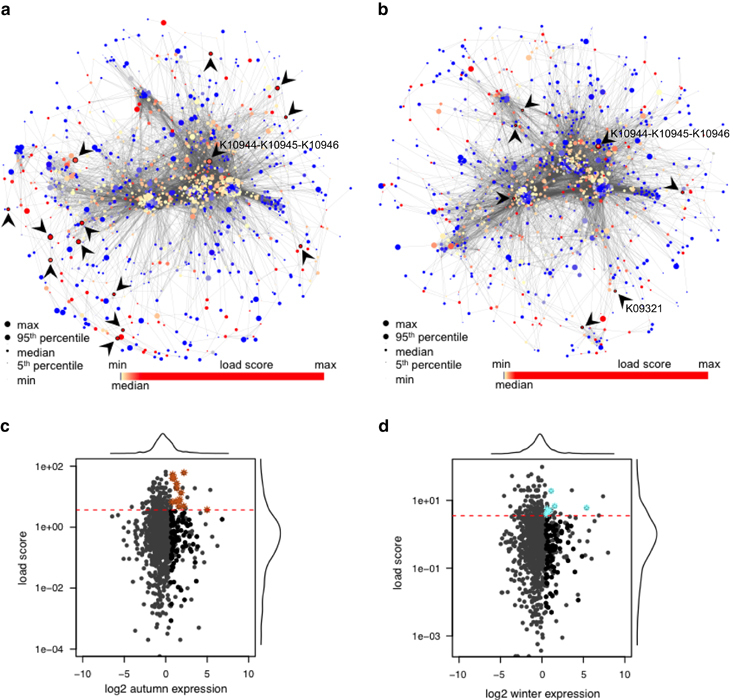
Methods: A comparative integrated omic analysis including metagenomics, metatranscriptomics and metaproteomics was carried out to elucidate functional differences between seasonally distinct oleaginous mixed microbial communities (OMMCs) sampled from an anoxic BWWT tank. A computational framework for the reconstruction of community-wide metabolic networks from multi-omic data was developed. These provide an overview of the functional capabilities by incorporating gene copy, transcript and protein abundances. To identify functional genes, which have a disproportionately important role in community function, we define a high relative gene expression and a high betweenness centrality relative to node degree as gene-centric and network topological features, respectively.
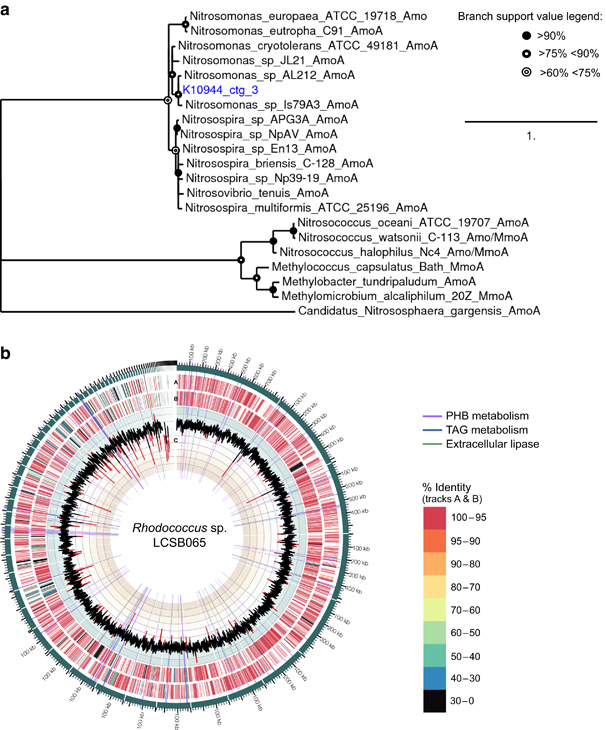
Results: Genes exhibiting high expression relative to gene copy abundance include genes involved in glycerolipid metabolism, particularly triacylglycerol lipase, encoded by known lipid accumulating populations, e.g., CandidatusMicrothrix parvicella. Genes with a high relative gene expression and topologically important positions in the network include genes involved in nitrogen metabolism and fatty acid biosynthesis, encoded by Nitrosomonas spp. and Rhodococcus spp. Such genes may be regarded as 'keystone genes' as they are likely to be encoded by keystone species.
Conclusion: The linking of key functionalities to community members through integrated omics opens up exciting possibilities for devising prediction and control strategies for microbial communities in the future.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Muller EE, Glaab E, May P, Vlassis N, Wilmes P. Condensing the omics fog of microbial communities. Trends Microbiol 2013; 7: 325–333. - PubMed
-
- Wilmes P, Bond PL. Microbial community proteomics: elucidating the catalysts and metabolic mechanisms that drive the Earth's biogeochemical cycles. Curr Opin Microbiol 2009; 12: 310–317. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
