

Front-End Electron Transfer Dissociation Coupled to a 21 Tesla FT-ICR Mass Spectrometer for Intact Protein Sequence Analysis
- PMID: 28721671
- PMCID: PMC5711562
- DOI: 10.1007/s13361-017-1702-3
Front-End Electron Transfer Dissociation Coupled to a 21 Tesla FT-ICR Mass Spectrometer for Intact Protein Sequence Analysis
Abstract
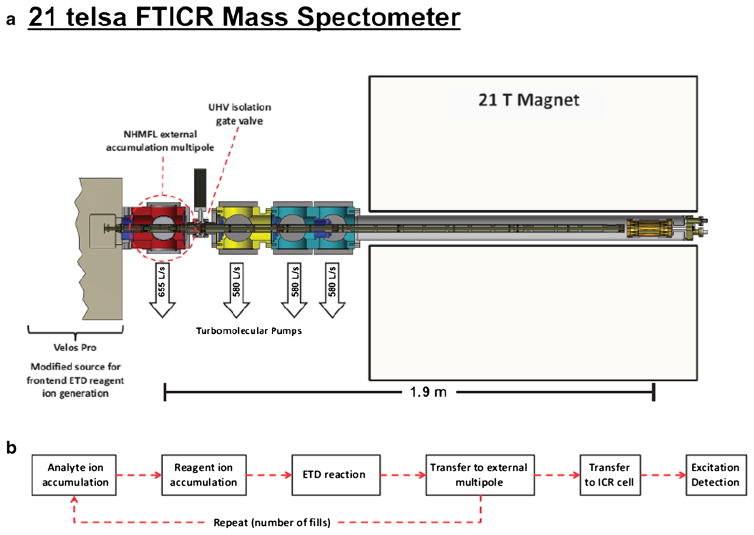
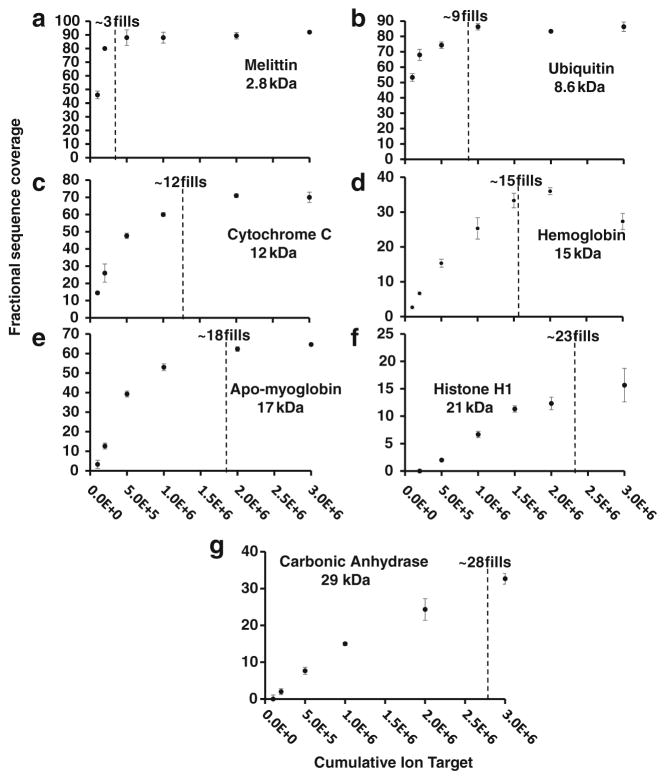
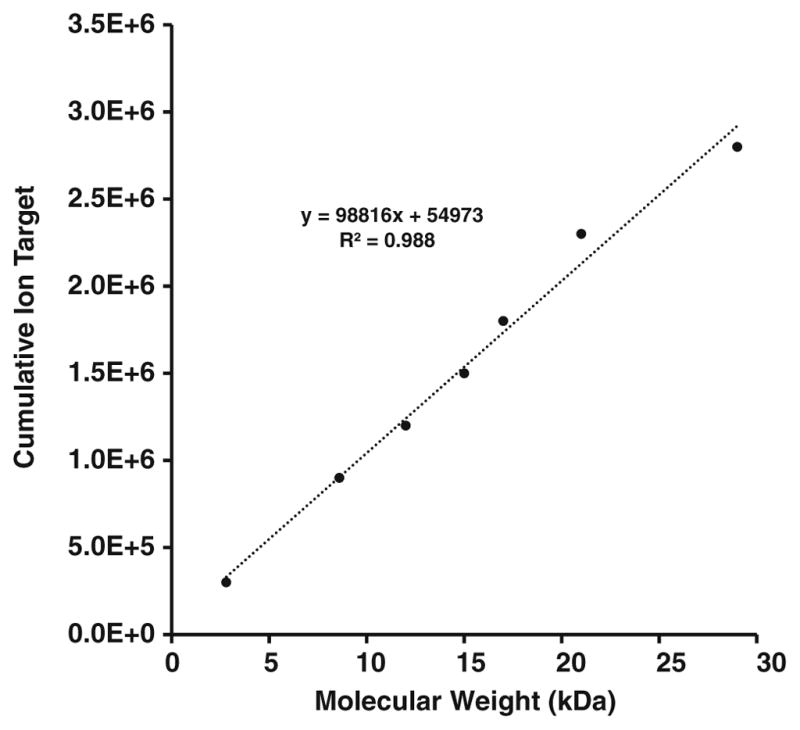
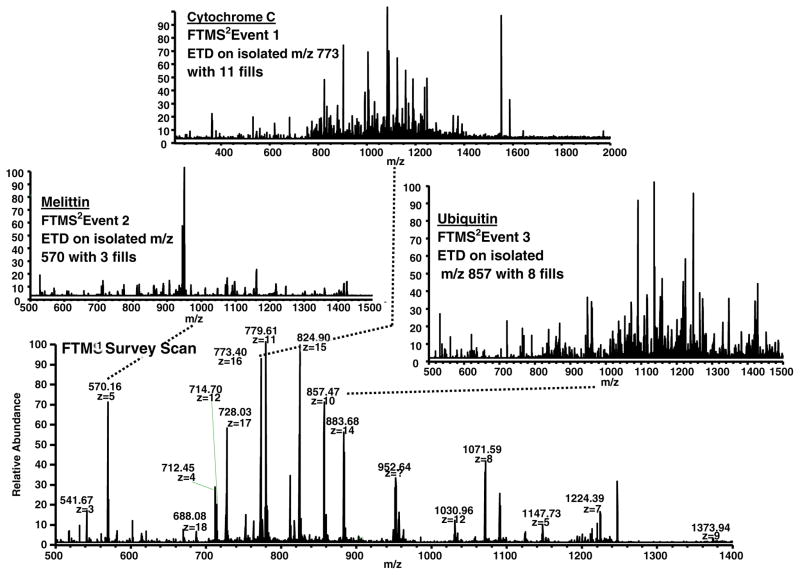
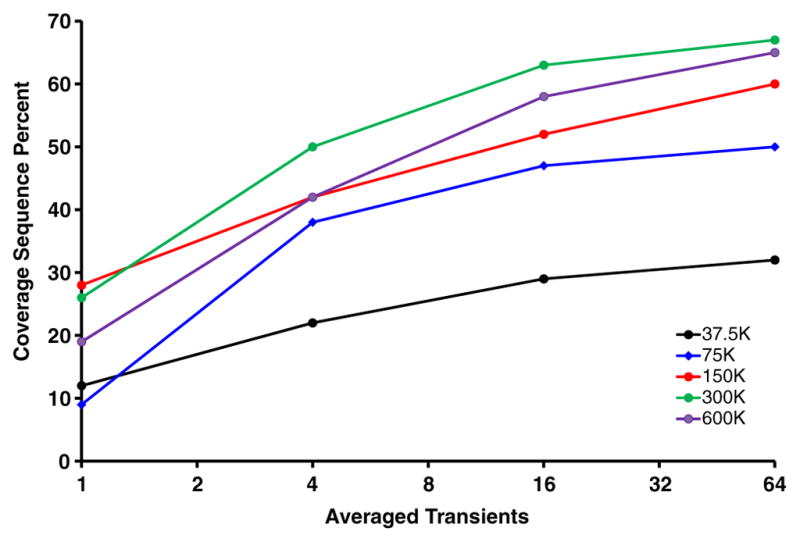
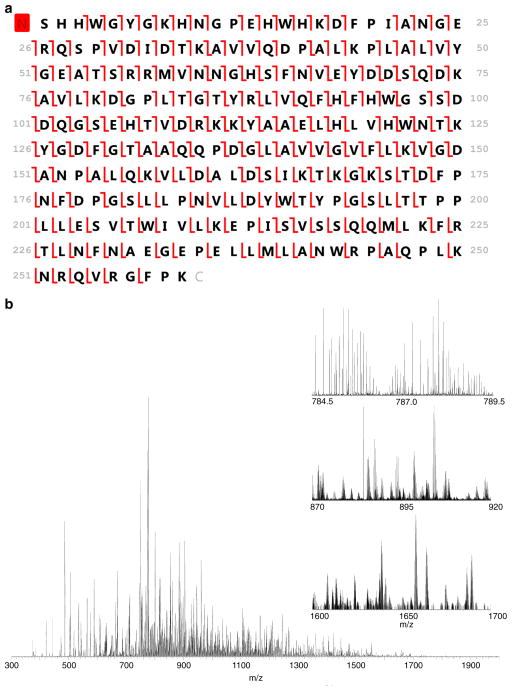
High resolution mass spectrometry is a key technology for in-depth protein characterization. High-field Fourier transform ion cyclotron resonance mass spectrometry (FT-ICR MS) enables high-level interrogation of intact proteins in the most detail to date. However, an appropriate complement of fragmentation technologies must be paired with FTMS to provide comprehensive sequence coverage, as well as characterization of sequence variants, and post-translational modifications. Here we describe the integration of front-end electron transfer dissociation (FETD) with a custom-built 21 tesla FT-ICR mass spectrometer, which yields unprecedented sequence coverage for proteins ranging from 2.8 to 29 kDa, without the need for extensive spectral averaging (e.g., ~60% sequence coverage for apo-myoglobin with four averaged acquisitions). The system is equipped with a multipole storage device separate from the ETD reaction device, which allows accumulation of multiple ETD fragment ion fills. Consequently, an optimally large product ion population is accumulated prior to transfer to the ICR cell for mass analysis, which improves mass spectral signal-to-noise ratio, dynamic range, and scan rate. We find a linear relationship between protein molecular weight and minimum number of ETD reaction fills to achieve optimum sequence coverage, thereby enabling more efficient use of instrument data acquisition time. Finally, real-time scaling of the number of ETD reactions fills during method-based acquisition is shown, and the implications for LC-MS/MS top-down analysis are discussed. Graphical Abstract ᅟ.
Keywords: ETD; Electrospray ionization; FTMS; Fourier transform mass spectrometry; Ion-ion reaction; MS/MS; MS2; Top-down.
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
