

Nanopore long-read RNAseq reveals widespread transcriptional variation among the surface receptors of individual B cells
- PMID: 28722025
- PMCID: PMC5524981
- DOI: 10.1038/ncomms16027
Nanopore long-read RNAseq reveals widespread transcriptional variation among the surface receptors of individual B cells
Abstract
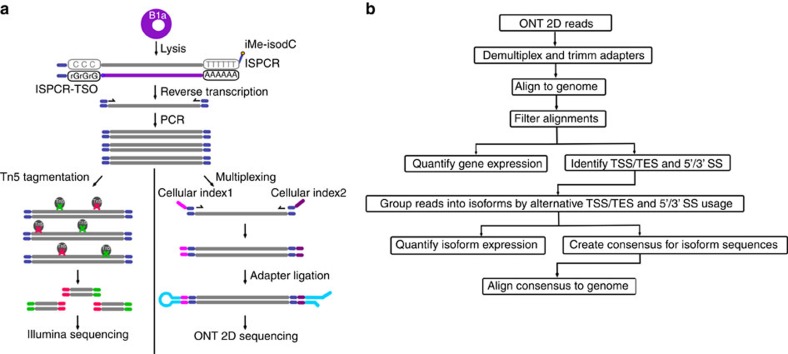
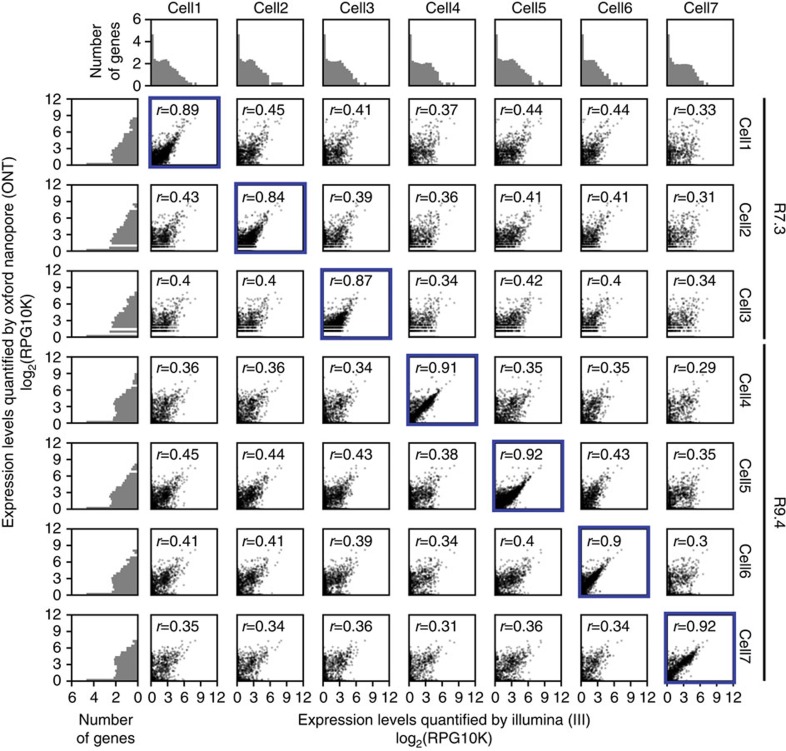
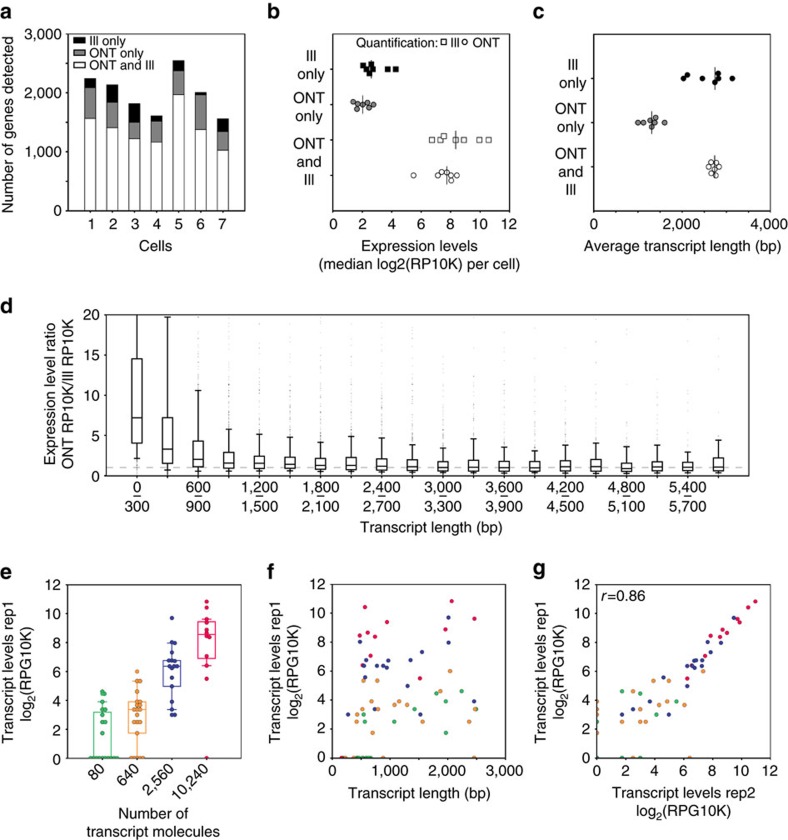
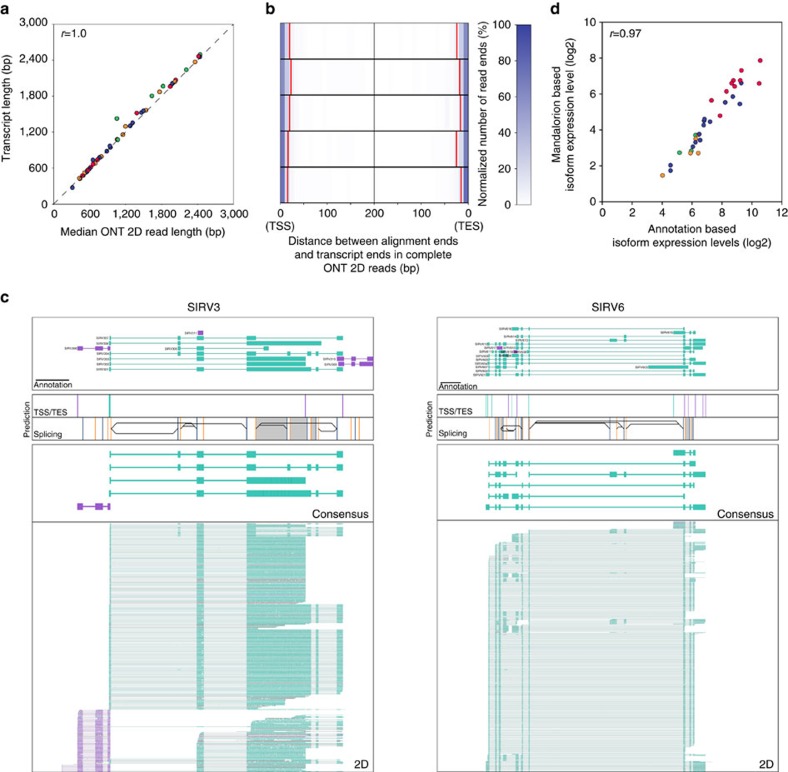
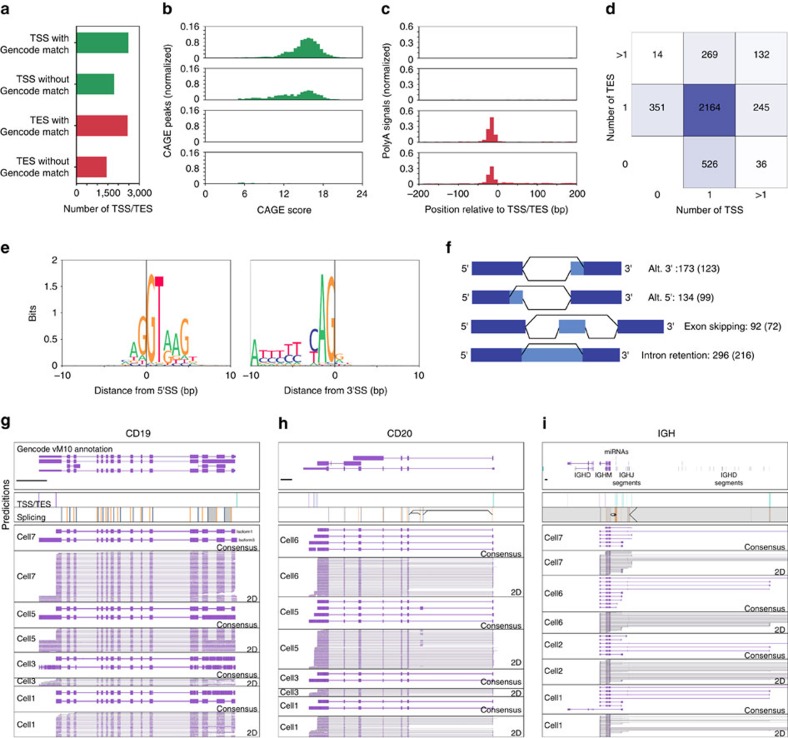
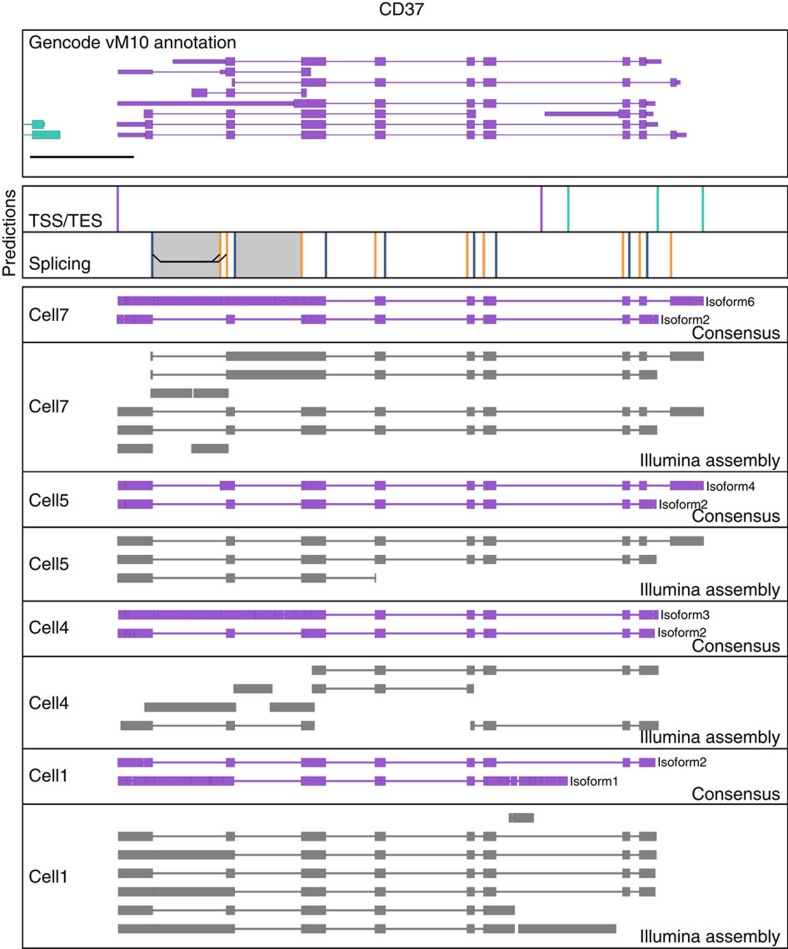
Understanding gene regulation and function requires a genome-wide method capable of capturing both gene expression levels and isoform diversity at the single-cell level. Short-read RNAseq is limited in its ability to resolve complex isoforms because it fails to sequence full-length cDNA copies of RNA molecules. Here, we investigate whether RNAseq using the long-read single-molecule Oxford Nanopore MinION sequencer is able to identify and quantify complex isoforms without sacrificing accurate gene expression quantification. After benchmarking our approach, we analyse individual murine B1a cells using a custom multiplexing strategy. We identify thousands of unannotated transcription start and end sites, as well as hundreds of alternative splicing events in these B1a cells. We also identify hundreds of genes expressed across B1a cells that display multiple complex isoforms, including several B cell-specific surface receptors. Our results show that we can identify and quantify complex isoforms at the single cell level.
Conflict of interest statement
M.A. is a paid consultant to Oxford Nanopore Technologies (Oxford, UK). The remaining authors declare no competing financial interests.
Figures
References
-
- Stamm S. et al. Function of alternative splicing. Gene 344, 1–20 (2005). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
