

Novel promoters and coding first exons in DLG2 linked to developmental disorders and intellectual disability
- PMID: 28724449
- PMCID: PMC5518101
- DOI: 10.1186/s13073-017-0452-y
Novel promoters and coding first exons in DLG2 linked to developmental disorders and intellectual disability
Abstract
Background: Tissue-specific integrative omics has the potential to reveal new genic elements important for developmental disorders.
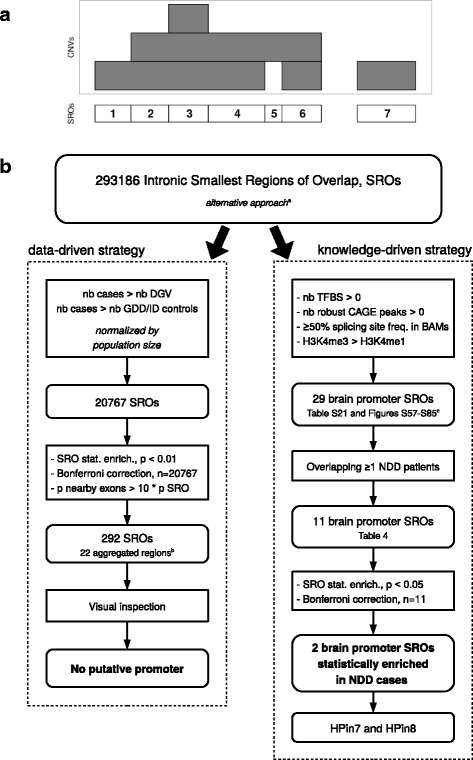
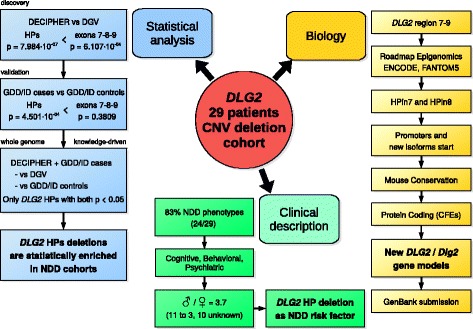
Methods: Two pediatric patients with global developmental delay and intellectual disability phenotype underwent array-CGH genetic testing, both showing a partial deletion of the DLG2 gene. From independent human and murine omics datasets, we combined copy number variations, histone modifications, developmental tissue-specific regulation, and protein data to explore the molecular mechanism at play.
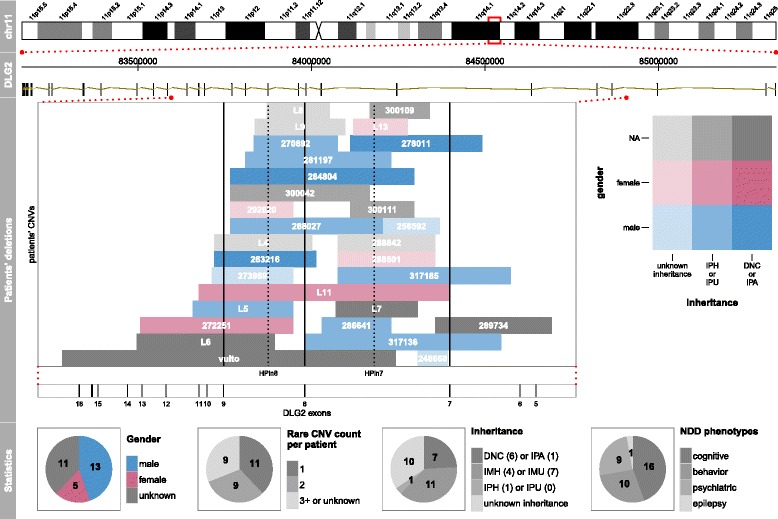
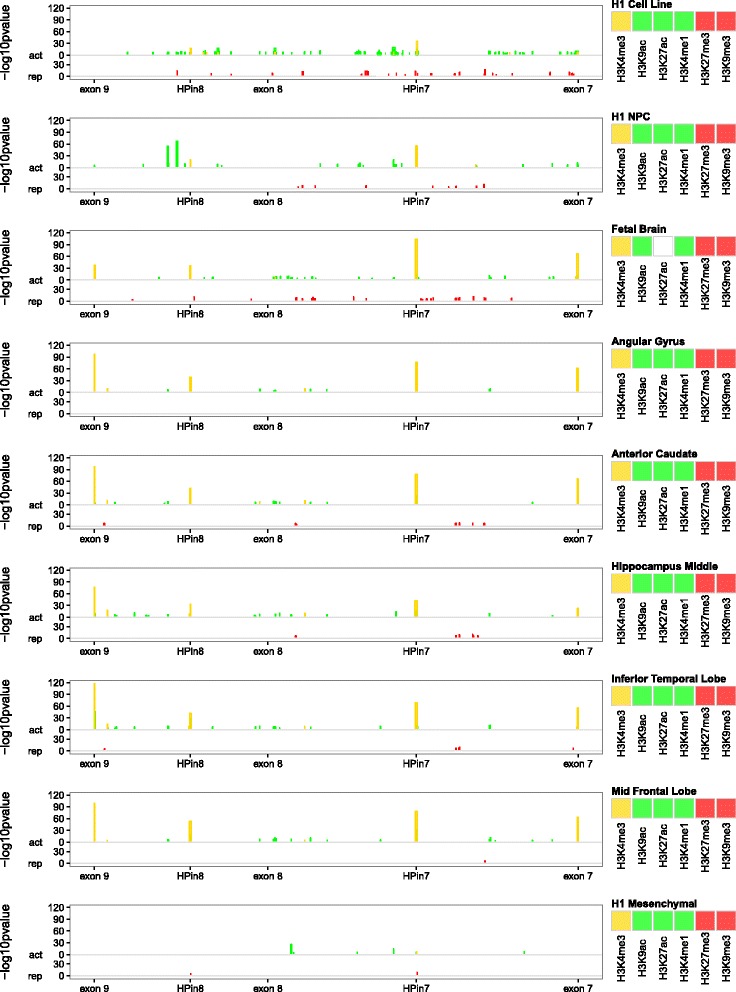
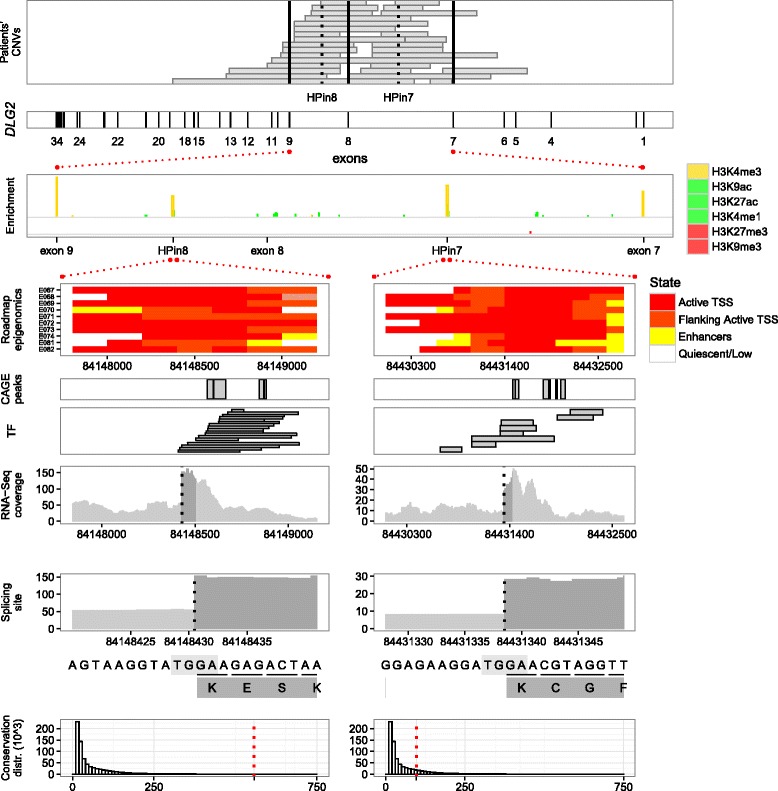
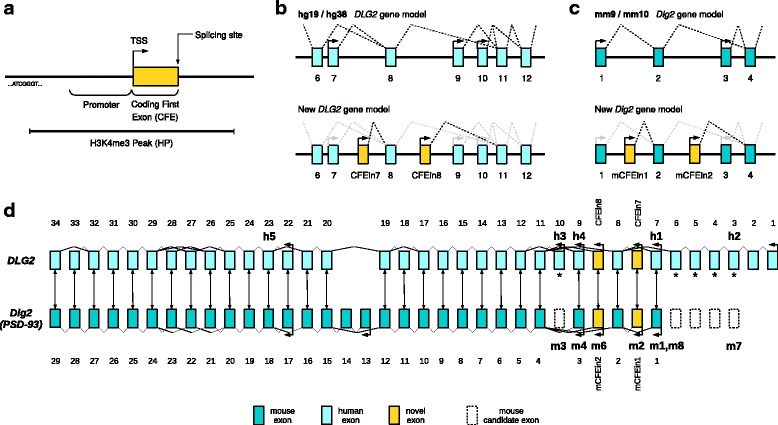
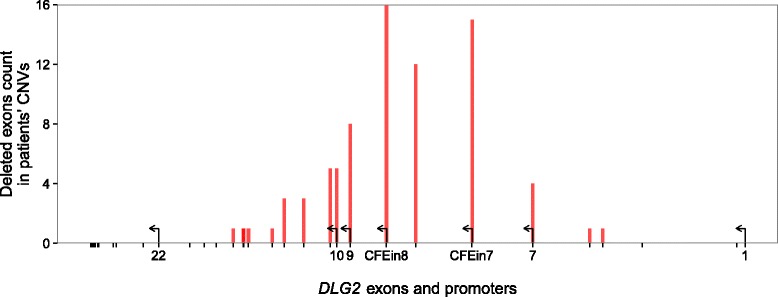
Results: Integrating genomics, transcriptomics, and epigenomics data, we describe two novel DLG2 promoters and coding first exons expressed in human fetal brain. Their murine conservation and protein-level evidence allowed us to produce new DLG2 gene models for human and mouse. These new genic elements are deleted in 90% of 29 patients (public and in-house) showing partial deletion of the DLG2 gene. The patients' clinical characteristics expand the neurodevelopmental phenotypic spectrum linked to DLG2 gene disruption to cognitive and behavioral categories.
Conclusions: While protein-coding genes are regarded as well known, our work shows that integration of multiple omics datasets can unveil novel coding elements. From a clinical perspective, our work demonstrates that two new DLG2 promoters and exons are crucial for the neurodevelopmental phenotypes associated with this gene. In addition, our work brings evidence for the lack of cross-annotation in human versus mouse reference genomes and nucleotide versus protein databases.
Keywords: DLG2; Functional genomics; Intellectual disability; Neurodevelopmental disorders; Promoters.
Conflict of interest statement
Consent for publication
The parents of patients 1 and 2 have provided consent to publish the clinical and genomic details presented.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
