

MUFINS: multi-formalism interaction network simulator
- PMID: 28725480
- PMCID: PMC5516860
- DOI: 10.1038/npjsba.2016.32
MUFINS: multi-formalism interaction network simulator
Abstract
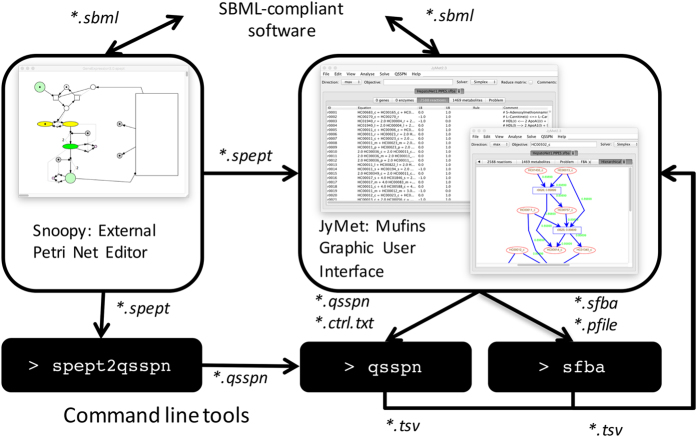
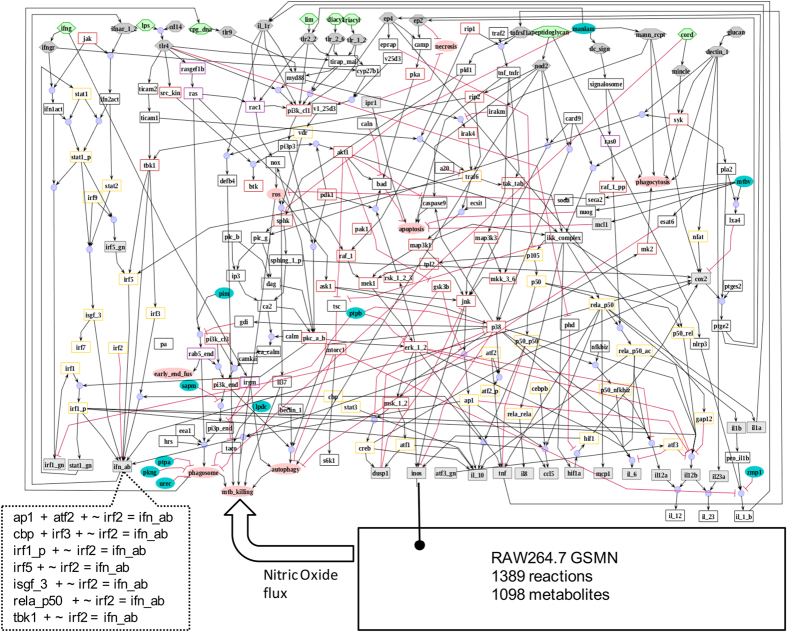
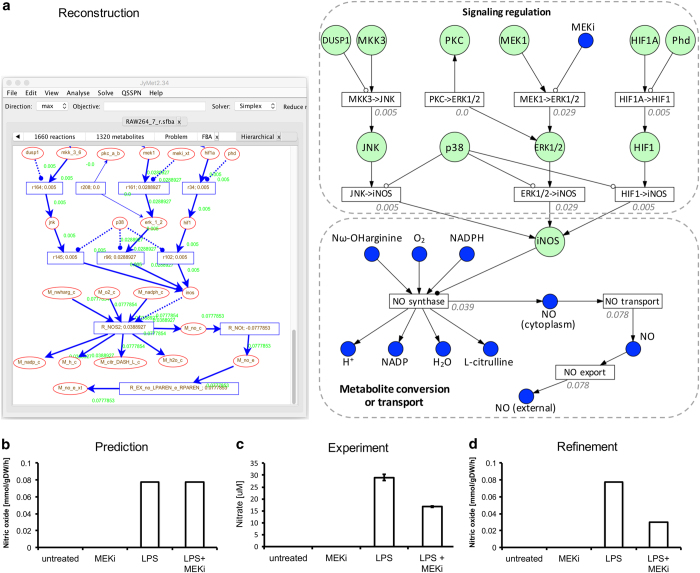
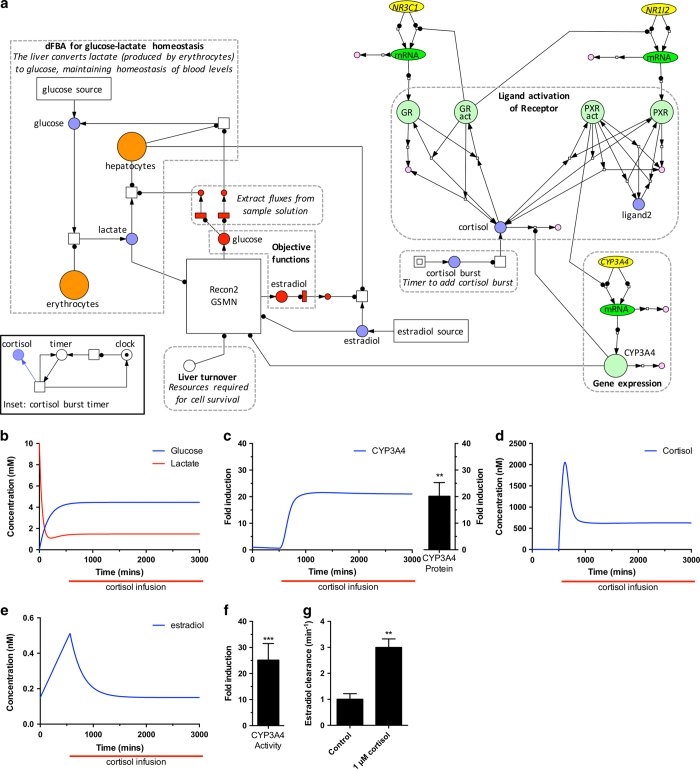
Systems Biology has established numerous approaches for mechanistic modeling of molecular networks in the cell and a legacy of models. The current frontier is the integration of models expressed in different formalisms to address the multi-scale biological system organization challenge. We present MUFINS (MUlti-Formalism Interaction Network Simulator) software, implementing a unique set of approaches for multi-formalism simulation of interaction networks. We extend the constraint-based modeling (CBM) framework by incorporation of linear inhibition constraints, enabling for the first time linear modeling of networks simultaneously describing gene regulation, signaling and whole-cell metabolism at steady state. We present a use case where a logical hypergraph model of a regulatory network is expressed by linear constraints and integrated with a Genome-Scale Metabolic Network (GSMN) of mouse macrophage. We experimentally validate predictions, demonstrating application of our software in an iterative cycle of hypothesis generation, validation and model refinement. MUFINS incorporates an extended version of our Quasi-Steady State Petri Net approach to integrate dynamic models with CBM, which we demonstrate through a dynamic model of cortisol signaling integrated with the human Recon2 GSMN and a model of nutrient dynamics in physiological compartments. Finally, we implement a number of methods for deriving metabolic states from ~omics data, including our new variant of the iMAT congruency approach. We compare our approach with iMAT through the analysis of 262 individual tumor transcriptomes, recovering features of metabolic reprogramming in cancer. The software provides graphics user interface with network visualization, which facilitates use by researchers who are not experienced in coding and mathematical modeling environments.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Gillespie, D. T. Exact stochastic simulation of coupled chemical-reactions. J. Phys. Chem. 81, 2340–2361 (1977).
-
- Tyson, J. J., Chen, K. & Novak, B. Network dynamics and cell physiology. Nat. Rev. Mol. Cell Biol. 2, 908–916 (2001). - PubMed
-
- Bordbar, A. et al. Constraint-based models predict metabolic and associated cellular functions. Nat. Rev. Genet. 15, 107–120 (2014). - PubMed
Grants and funding
- BB/I004696/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/I00453X/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/I004688/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/D019079/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/F003552/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/C008219/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/F003528/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/I00470X/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/F003536/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/F003544/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
