

Systems biology driving drug development: from design to the clinical testing of the anti-ErbB3 antibody seribantumab (MM-121)
- PMID: 28725482
- PMCID: PMC5516865
- DOI: 10.1038/npjsba.2016.34
Systems biology driving drug development: from design to the clinical testing of the anti-ErbB3 antibody seribantumab (MM-121)
Abstract
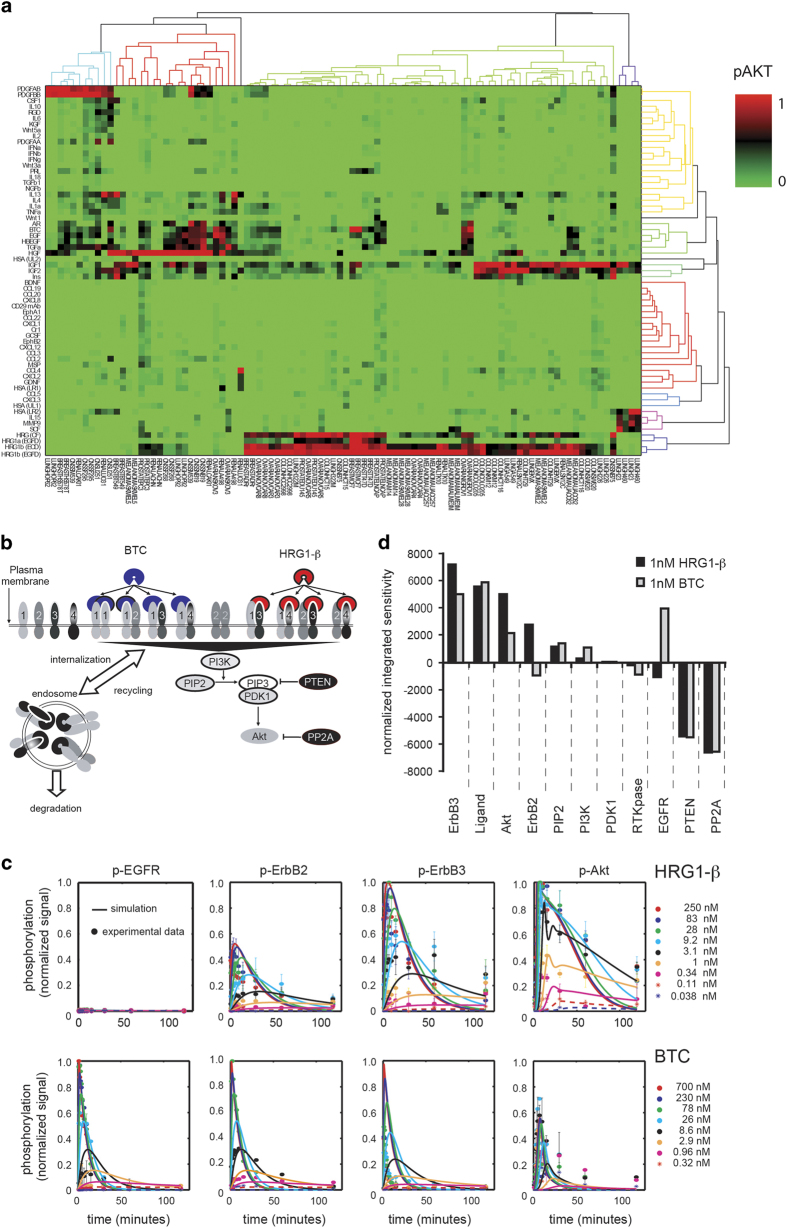
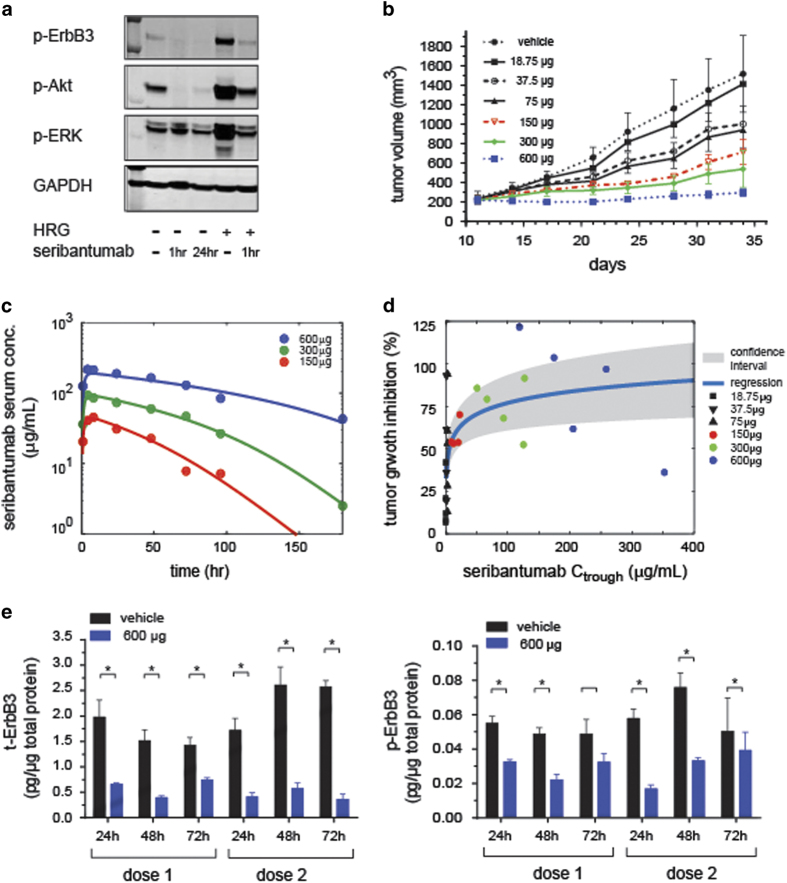
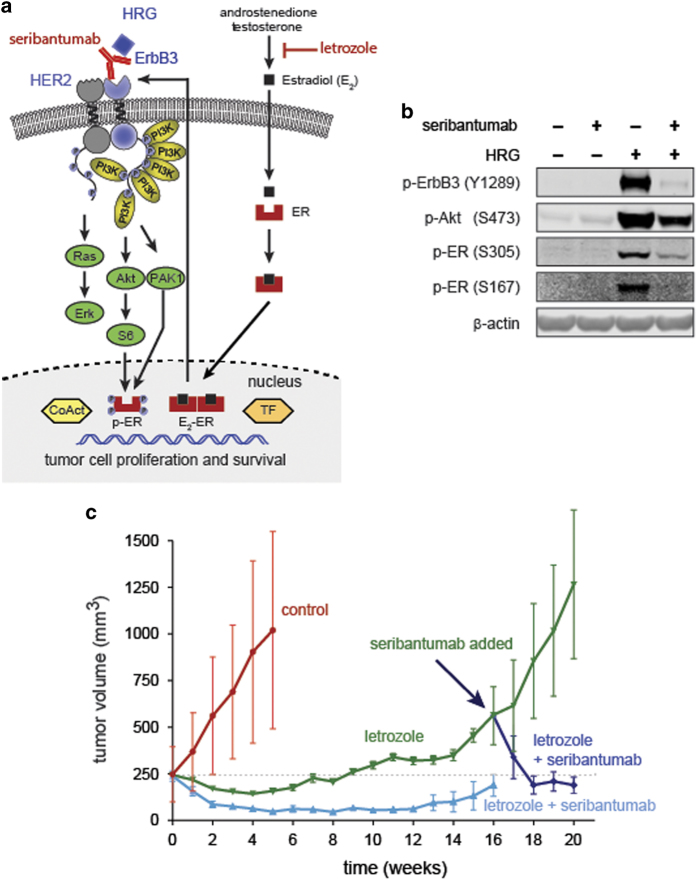
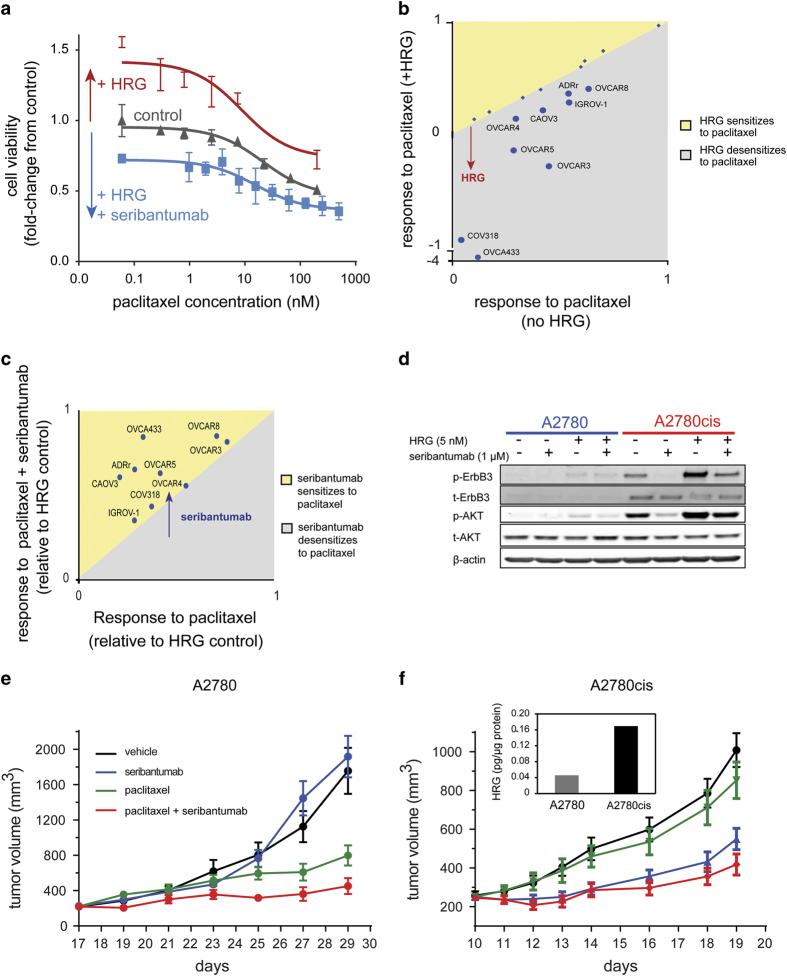
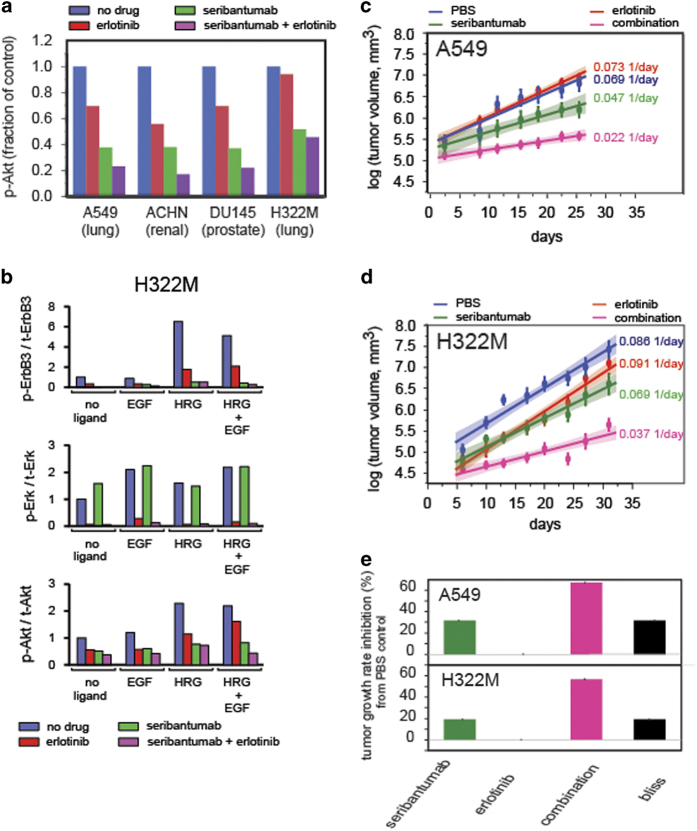
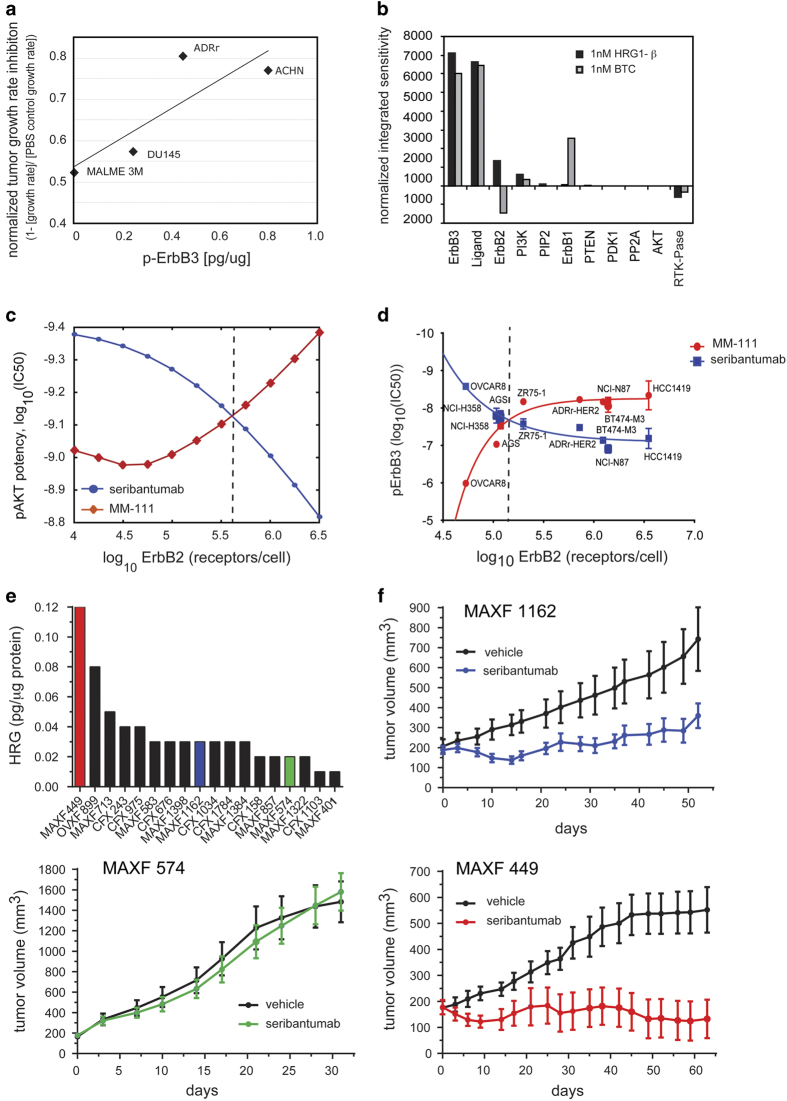
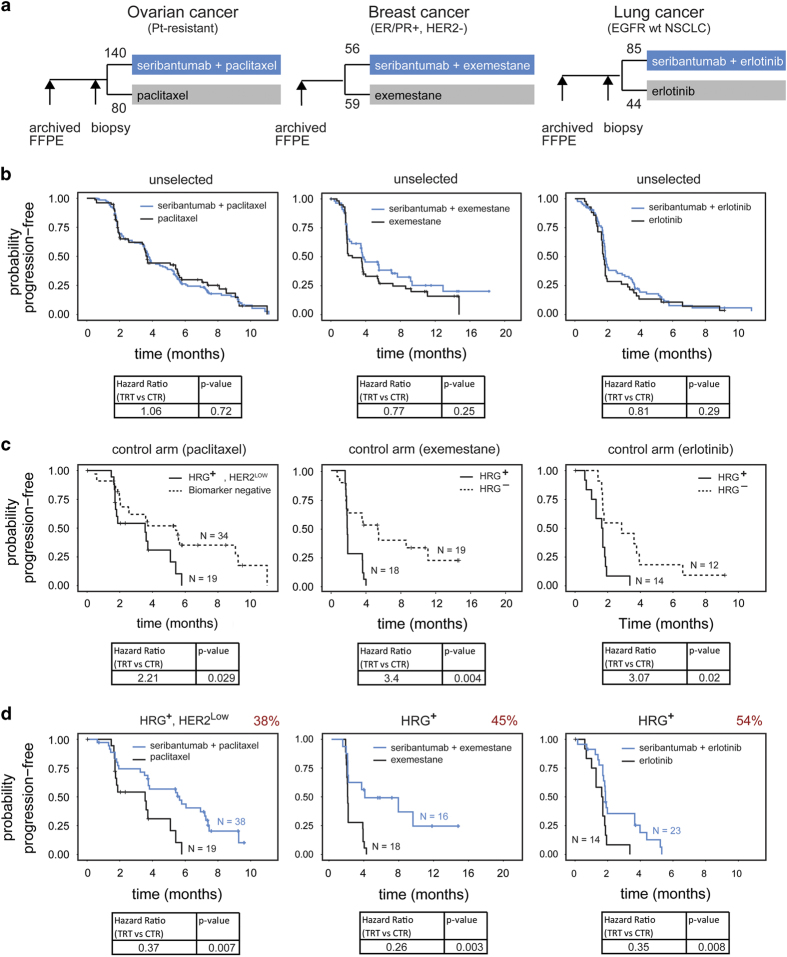
The ErbB family of receptor tyrosine kinases comprises four members: epidermal growth factor receptor (EGFR/ErbB1), human EGFR 2 (HER2/ErbB2), ErbB3/HER3, and ErbB4/HER4. The first two members of this family, EGFR and HER2, have been implicated in tumorigenesis and cancer progression for several decades, and numerous drugs have now been approved that target these two proteins. Less attention, however, has been paid to the role of this family in mediating cancer cell survival and drug tolerance. To better understand the complex signal transduction network triggered by the ErbB receptor family, we built a computational model that quantitatively captures the dynamics of ErbB signaling. Sensitivity analysis identified ErbB3 as the most critical activator of phosphoinositide 3-kinase (PI3K) and Akt signaling, a key pro-survival pathway in cancer cells. Based on this insight, we designed a fully human monoclonal antibody, seribantumab (MM-121), that binds to ErbB3 and blocks signaling induced by the extracellular growth factors heregulin (HRG) and betacellulin (BTC). In this article, we present some of the key preclinical simulations and experimental data that formed the scientific foundation for three Phase 2 clinical trials in metastatic cancer. These trials were designed to determine if patients with advanced malignancies would derive benefit from the addition of seribantumab to standard-of-care drugs in platinum-resistant/refractory ovarian cancer, hormone receptor-positive HER2-negative breast cancer, and EGFR wild-type non-small cell lung cancer (NSCLC). From preclinical studies we learned that basal levels of ErbB3 phosphorylation correlate with response to seribantumab monotherapy in mouse xenograft models. As ErbB3 is rapidly dephosphorylated and hence difficult to measure clinically, we used the computational model to identify a set of five surrogate biomarkers that most directly affect the levels of p-ErbB3: HRG, BTC, EGFR, HER2, and ErbB3. Preclinically, the combined information from these five markers was sufficient to accurately predict which xenograft models would respond to seribantumab, and the single-most accurate predictor was HRG. When tested clinically in ovarian, breast and lung cancer, HRG mRNA expression was found to be both potentially prognostic of insensitivity to standard therapy and potentially predictive of benefit from the addition of seribantumab to standard of care therapy in all three indications. In addition, it was found that seribantumab was most active in cancers with low levels of HER2, consistent with preclinical predictions. Overall, our clinical studies and studies of others suggest that HRG expression defines a drug-tolerant cancer cell phenotype that persists in most solid tumor indications and may contribute to rapid clinical progression. To our knowledge, this is the first example of a drug designed and clinically tested using the principles of Systems Biology.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Schoeberl, B. et al. Therapeutically targeting ErbB3: a key node in ligand-induced activation of the ErbB receptor-PI3K axis. Sci. Signal. 2, ra31 (2009). - PubMed
-
- Chabner, B. A. & Roberts, T. G. Timeline: Chemotherapy and the war on cancer. Nat. Rev. Cancer 5, 65–72 (2005). - PubMed
-
- Hudis, C. A. Trastuzumab--mechanism of action and use in clinical practice. N. Engl. J. Med. 357, 39–51 (2007). - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
