

Dynamics of genomic innovation in the unicellular ancestry of animals
- PMID: 28726632
- PMCID: PMC5560861
- DOI: 10.7554/eLife.26036
Dynamics of genomic innovation in the unicellular ancestry of animals
Abstract
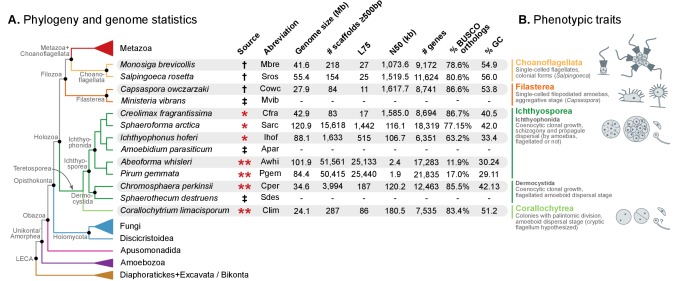
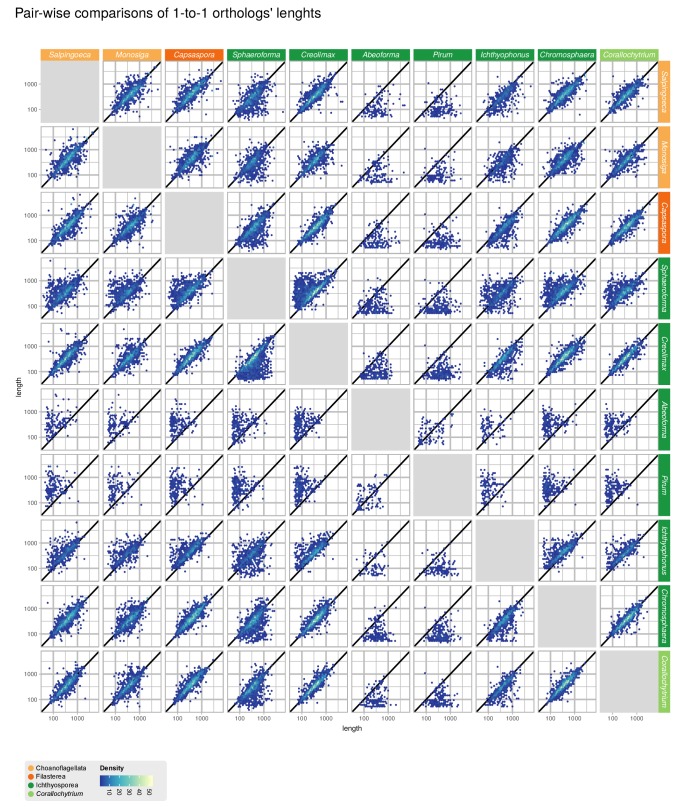
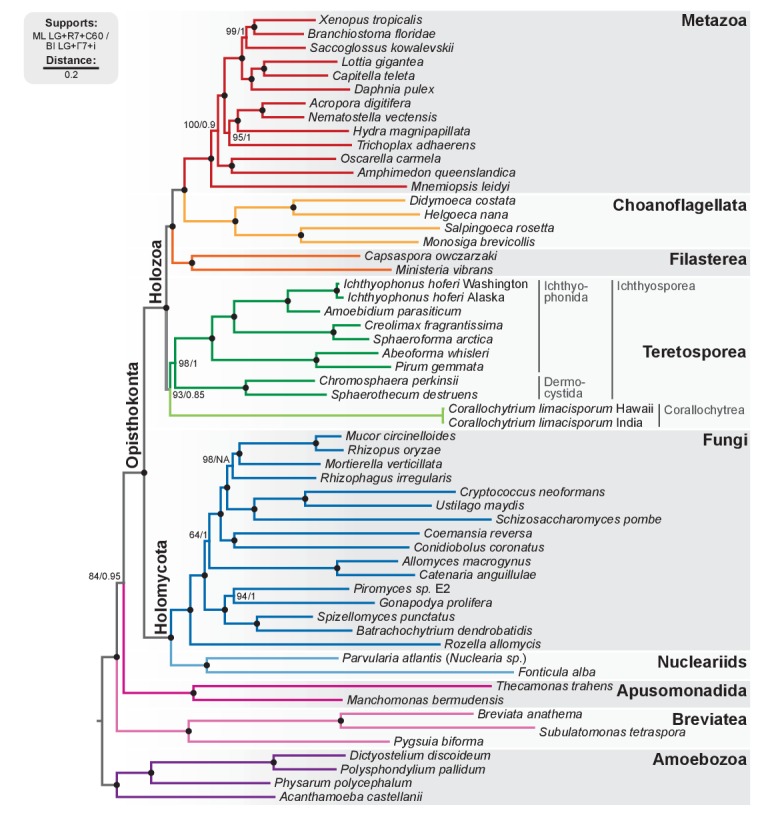
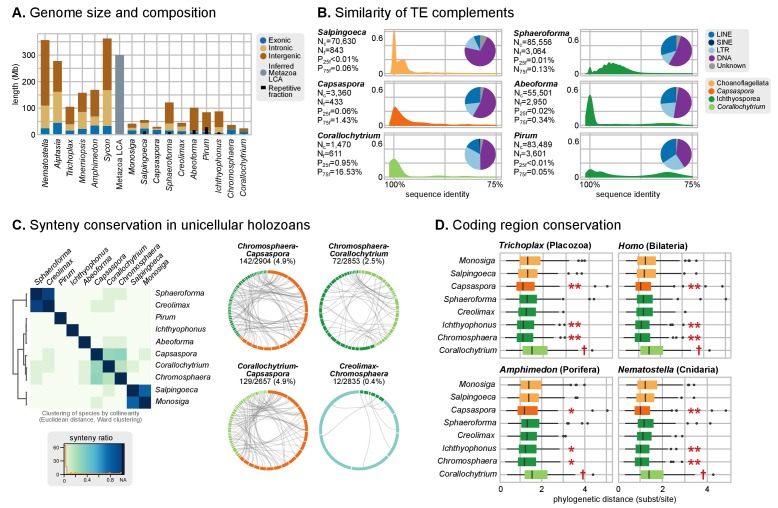
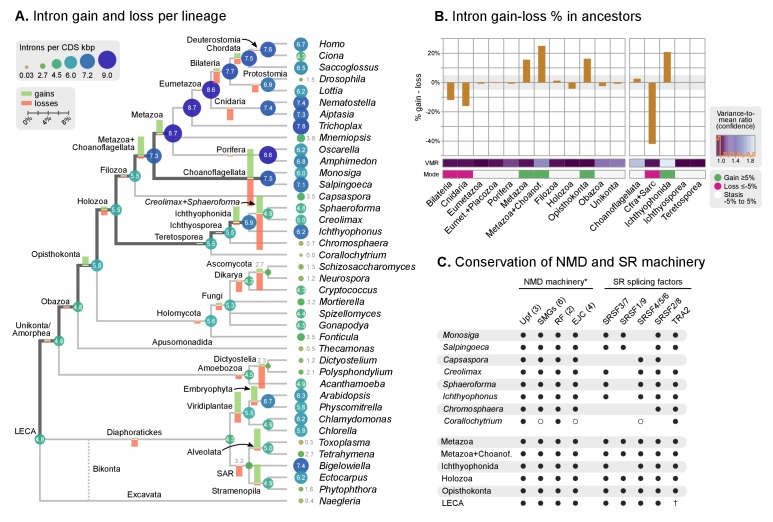
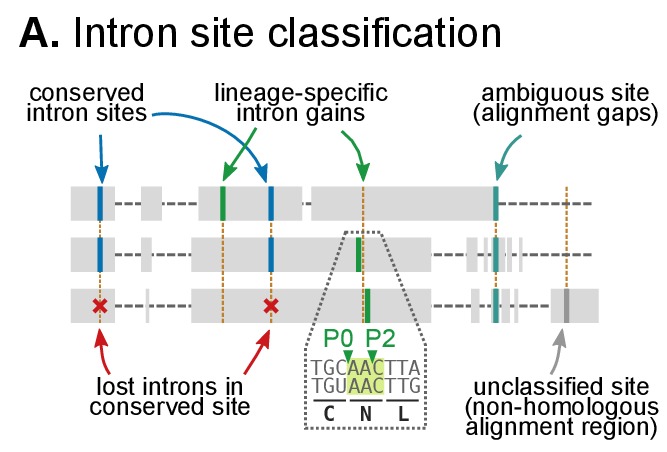
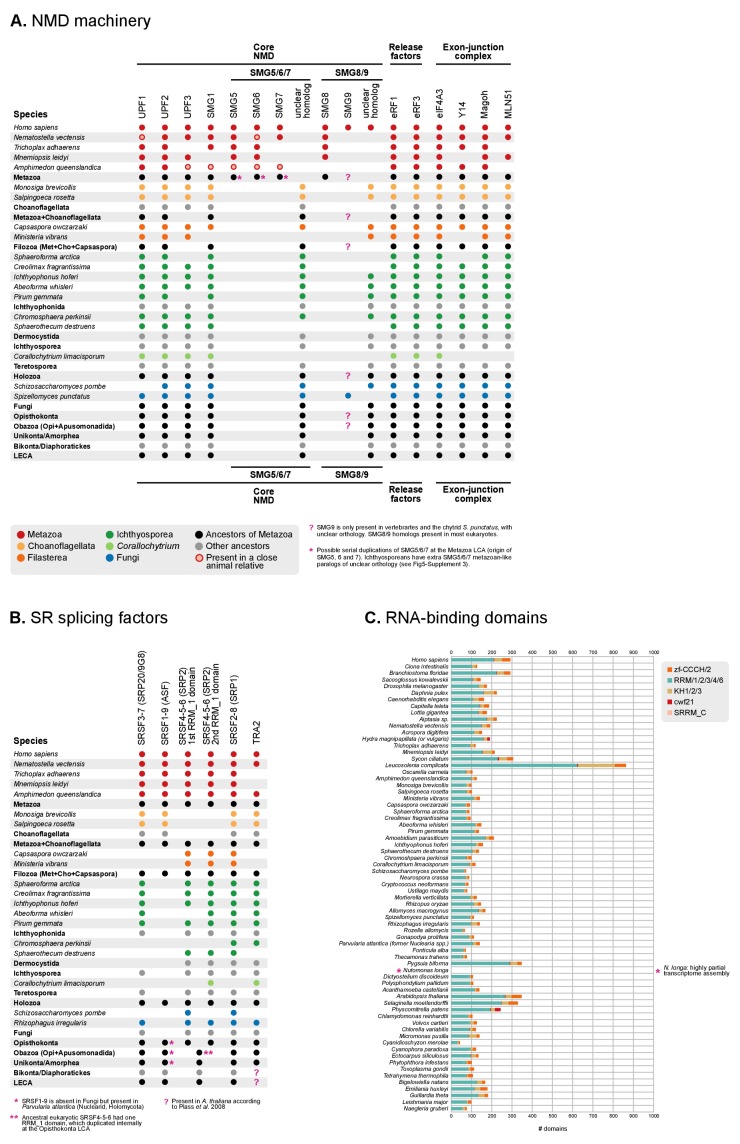
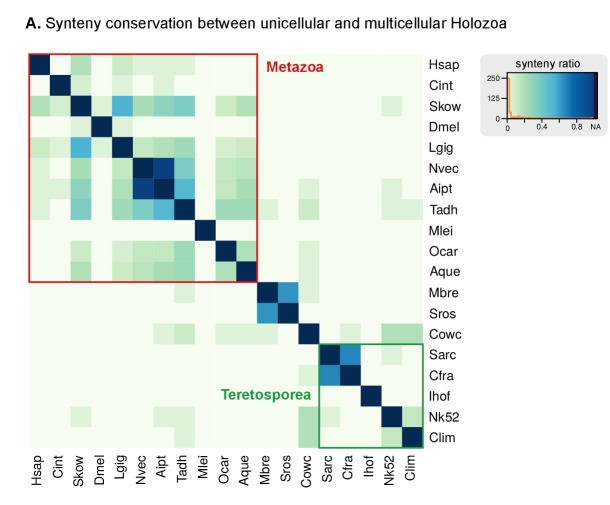
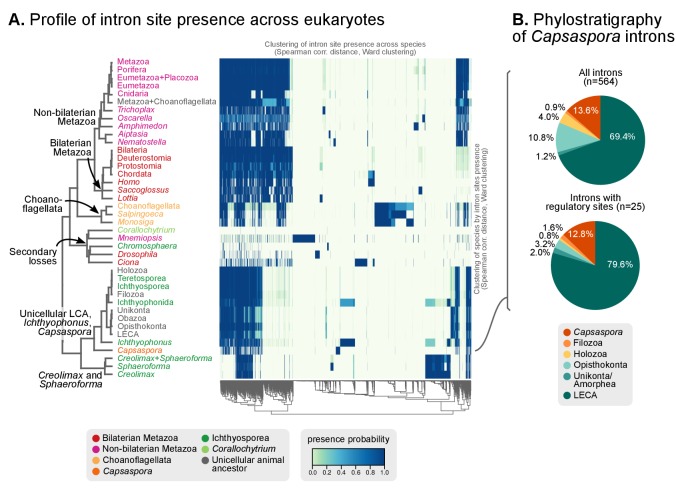
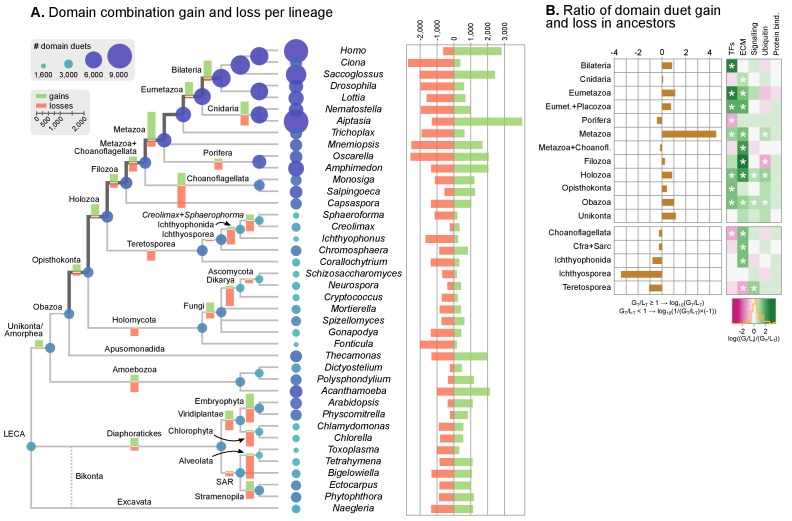
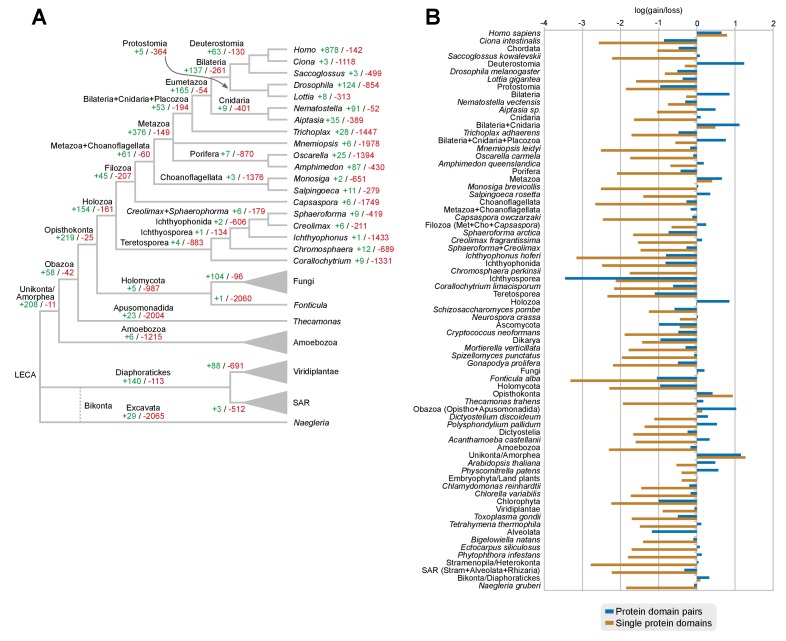
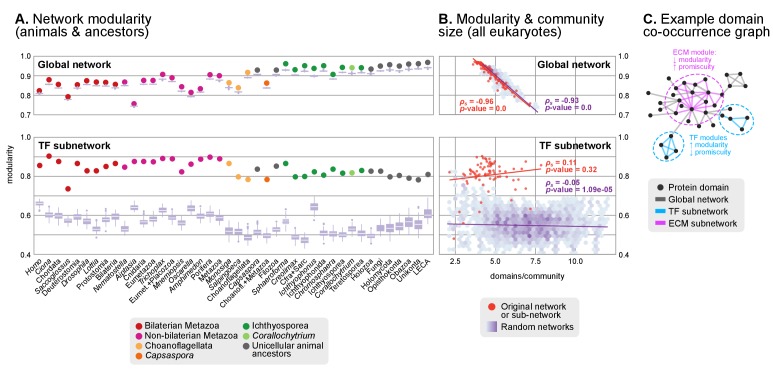
Which genomic innovations underpinned the origin of multicellular animals is still an open debate. Here, we investigate this question by reconstructing the genome architecture and gene family diversity of ancestral premetazoans, aiming to date the emergence of animal-like traits. Our comparative analysis involves genomes from animals and their closest unicellular relatives (the Holozoa), including four new genomes: three Ichthyosporea and Corallochytrium limacisporum. Here, we show that the earliest animals were shaped by dynamic changes in genome architecture before the emergence of multicellularity: an early burst of gene diversity in the ancestor of Holozoa, enriched in transcription factors and cell adhesion machinery, was followed by multiple and differently-timed episodes of synteny disruption, intron gain and genome expansions. Thus, the foundations of animal genome architecture were laid before the origin of complex multicellularity - highlighting the necessity of a unicellular perspective to understand early animal evolution.
Keywords: animal origins; chromosomes; comparative genomics; evolutionary biology; gene family evolution; genes; genomics; holozoans; ichthyosporeans; introns.
Conflict of interest statement
The authors declare that no competing interests exist.
Figures

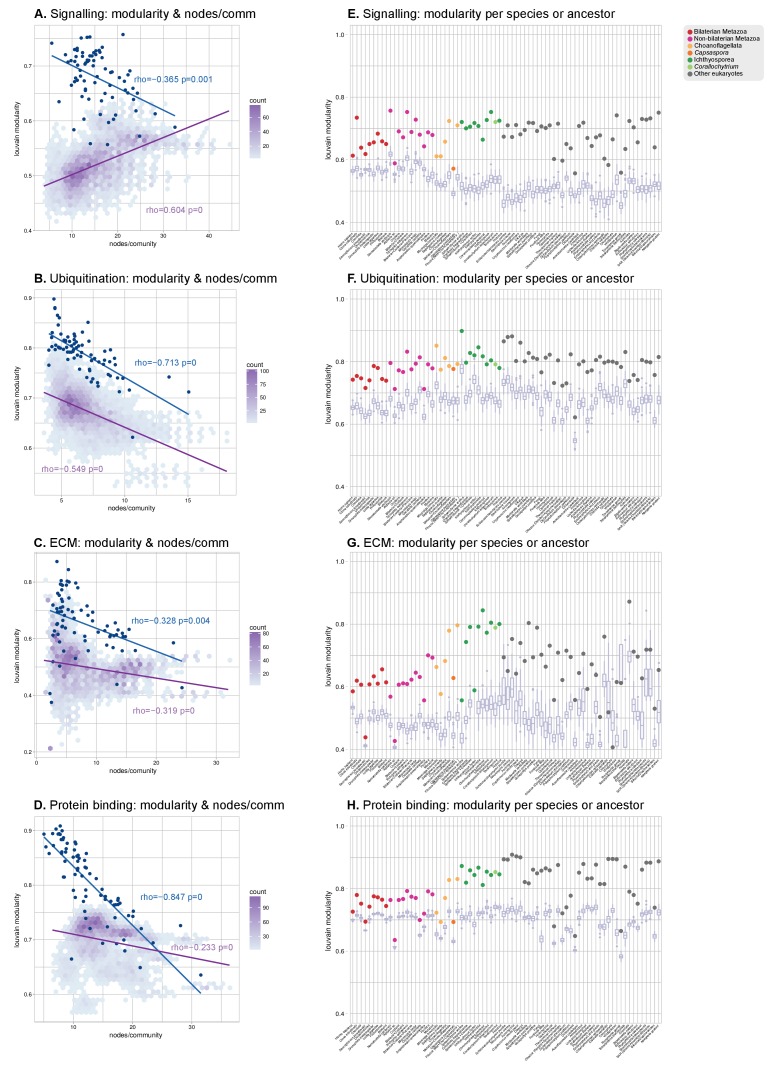
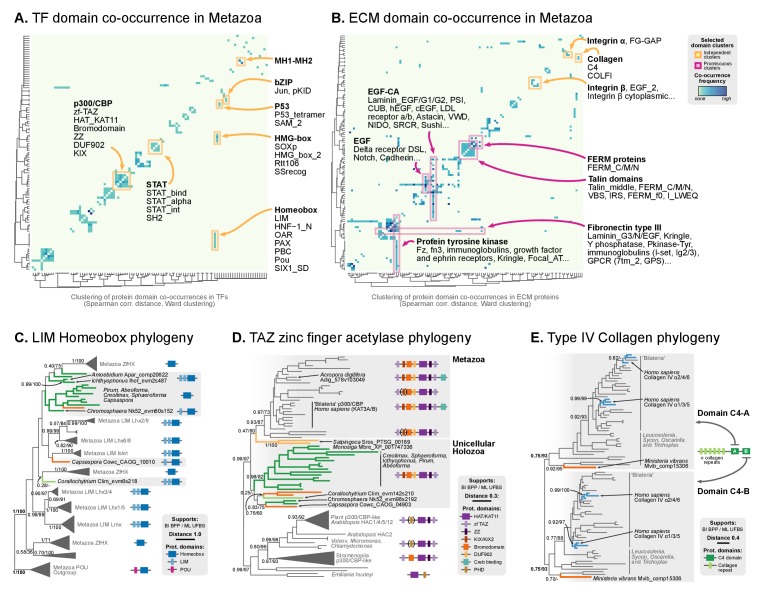
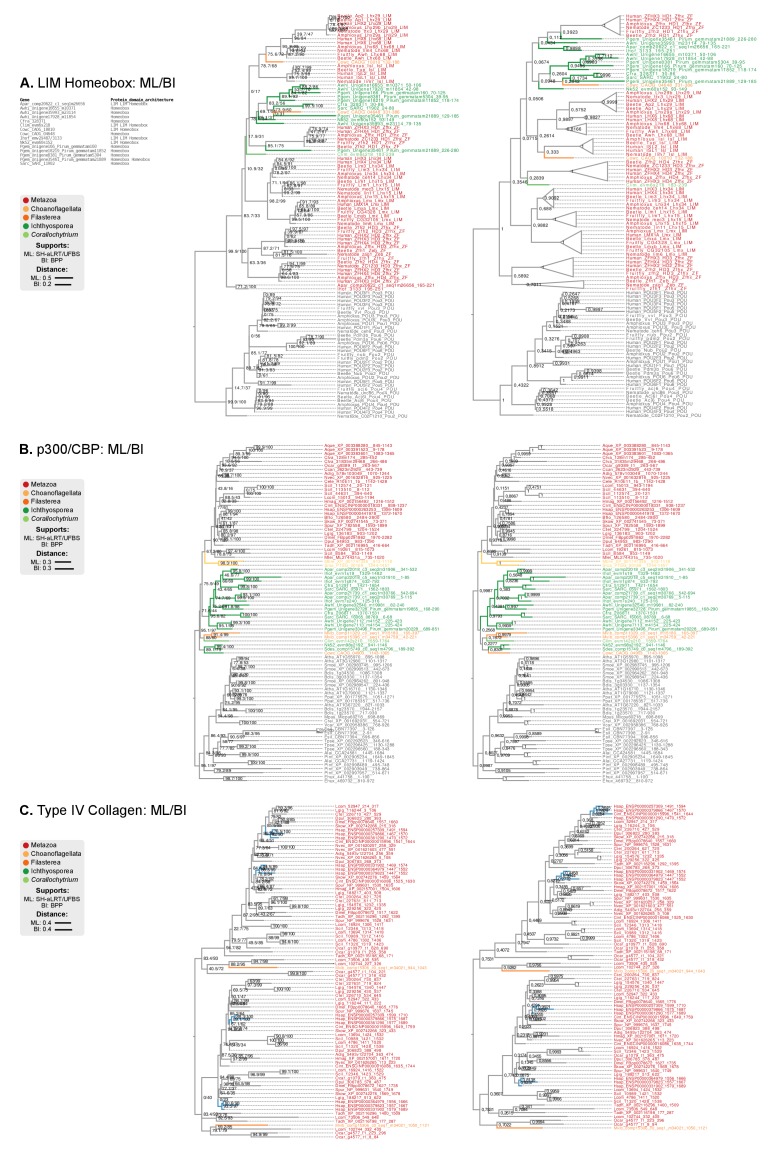
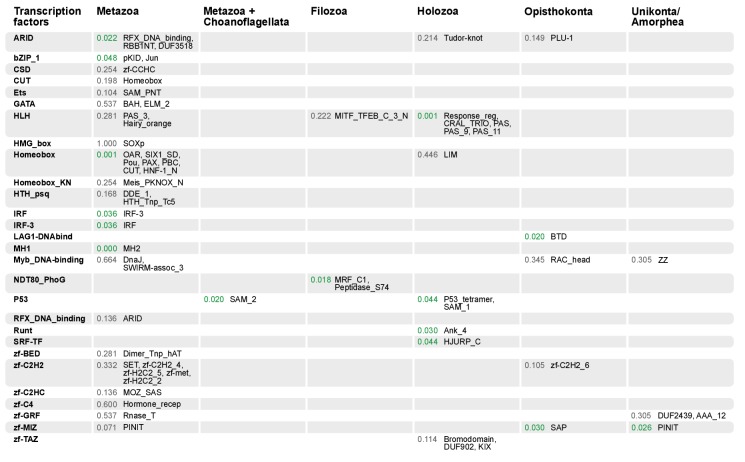
References
-
- Andrews S. FastQC. [3 May, 2016];2014 http://www.bioinformatics.bbsrc.ac.uk/projects/fastqc
-
- Aouacheria A, Geourjon C, Aghajari N, Navratil V, Deléage G, Lethias C, Exposito JY. Insights into early extracellular matrix evolution: spongin short chain collagen-related proteins are homologous to basement membrane type IV collagens and form a novel family widely distributed in invertebrates. Molecular Biology and Evolution. 2006;23:2288–2302. doi: 10.1093/molbev/msl100. - DOI - PubMed
-
- Barbosa-Morais NL, Irimia M, Pan Q, Xiong HY, Gueroussov S, Lee LJ, Slobodeniuc V, Kutter C, Watt S, Colak R, Kim T, Misquitta-Ali CM, Wilson MD, Kim PM, Odom DT, Frey BJ, Blencowe BJ. The evolutionary landscape of alternative splicing in vertebrate species. Science. 2012;338:1587–1593. doi: 10.1126/science.1230612. - DOI - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
