

Mapping genome-wide transcription-factor binding sites using DAP-seq
- PMID: 28726847
- PMCID: PMC5576341
- DOI: 10.1038/nprot.2017.055
Mapping genome-wide transcription-factor binding sites using DAP-seq
Abstract
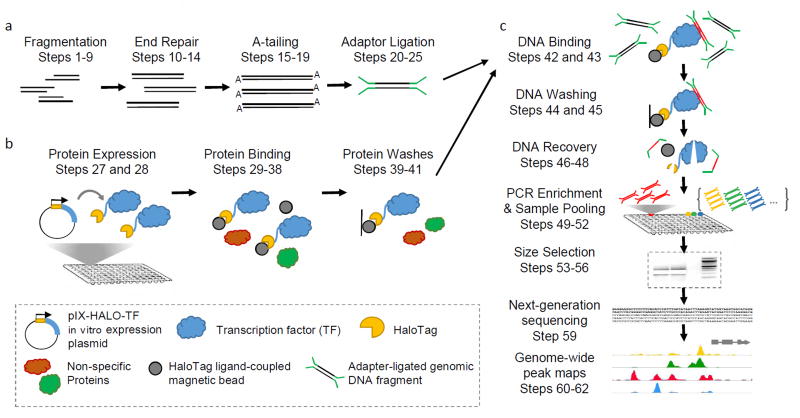
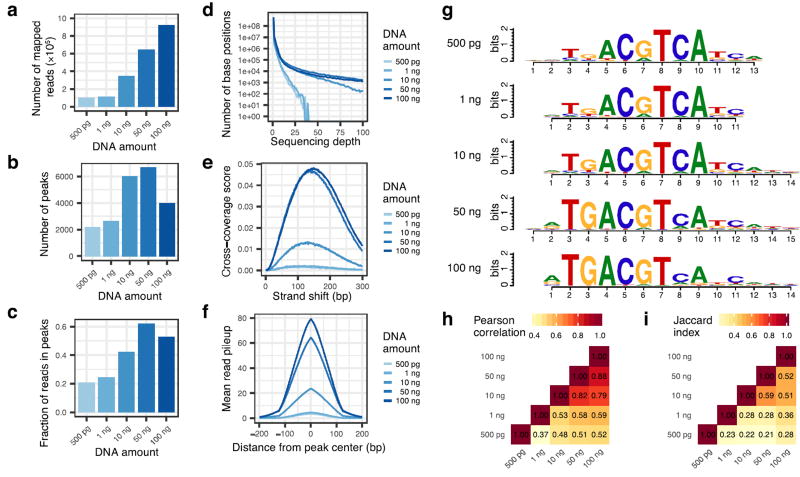
To enable low-cost, high-throughput generation of cistrome and epicistrome maps for any organism, we developed DNA affinity purification sequencing (DAP-seq), a transcription factor (TF)-binding site (TFBS) discovery assay that couples affinity-purified TFs with next-generation sequencing of a genomic DNA library. The method is fast, inexpensive, and more easily scaled than chromatin immunoprecipitation sequencing (ChIP-seq). DNA libraries are constructed using native genomic DNA from any source of interest, preserving cell- and tissue-specific chemical modifications that are known to affect TF binding (such as DNA methylation) and providing increased specificity as compared with in silico predictions based on motifs from methods such as protein-binding microarrays (PBMs) and systematic evolution of ligands by exponential enrichment (SELEX). The resulting DNA library is incubated with an affinity-tagged in vitro-expressed TF, and TF-DNA complexes are purified using magnetic separation of the affinity tag. Bound genomic DNA is eluted from the TF and sequenced using next-generation sequencing. Sequence reads are mapped to a reference genome, identifying genome-wide binding locations for each TF assayed, from which sequence motifs can then be derived. A researcher with molecular biology experience should be able to follow this protocol, processing up to 400 samples per week.
Conflict of interest statement
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
Figures
References
-
- Babu MM, Luscombe NM, 3, Aravind L, Gerstein M, Teichmann SA. Structure and evolution of transcriptional regulatory networks. Curr. Opin. Struct. Biol. 2004;14:283–291. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
